Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Lymphopoiesis wikipedia , lookup
Hygiene hypothesis wikipedia , lookup
Adaptive immune system wikipedia , lookup
Molecular mimicry wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Pathophysiology of multiple sclerosis wikipedia , lookup
Ulcerative colitis wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Innate immune system wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
Sjögren syndrome wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Marleen I. Verstege
Epithelial barrier and dendritic cell function in the intestinal mucosa
2010
ISBN: 978-90-8570-566-6
Epithelial barrier
and dendritic cell
function in the
intestinal mucosa
Marleen I. Verstege
Epithelial barrier and dendritic cell
function in the intestinal mucosa
Cover design and layout: Marleen Verstege, de foto’s op de cover en binnenzijde zijn
genomen in de Kaapse bossen bij Doorn in de herfst van 2009.
ISBN: 978-90-8570-566-6
Print: Wöhrmann Print Service, Zutphen
© 2010 Marleen I. Verstege, Maarssen, The Netherlands
Epithelial barrier and dendritic cell
function in the intestinal mucosa
ACADEMISCH PROEFSCHRIFT
ter verkrijging van de graad doctor
aan de Universtiteit van Amsterdam,
op gezag van de Rector Magnificus
prof. dr. D.C. van den Boom
ten overstaan van een door het college voor promoties
ingestelde promotiecommissie,
in het openbaar te verdedigen in de Agnietenkapel
op dinsdag 29 juni 2010, te 14:00 uur
door
Marleen Ingeborg Verstege
geboren te Maarssen
Promotiecommissie:
Promotor:
Prof. Dr. G.E. Boeckxstaens
Co-promotoren:
Dr. A.A. te Velde
Dr. W.J. de Jonge
Overige leden:
Prof.dr. H. Spits
Prof.dr. M.L.Kapsenberg
Prof.dr. C.J.F. van Noorden
Prof.dr. Y. van Kooyk
Prof.dr. F.J.W. ten Kate
Prof.dr. D.W. Hommes
Dr. F.A. Vyth-Dreese
Faculteit der Geneeskunde
Aan een ieder die een bijdrage heeft geleverd aan dit proefschrift
To all who contributed to this thesis
Na een lange dag hard werken
Loop ik opgewekt naar buiten.
Ik zie het zonnetje stralend schijnen
En ik hoor de vogeltjes vrolijk fluiten.
Ik ben op weg naar huis: naar mijn vriend en kind,
Terwijl zich in mijn buik nog een hartje bevindt.
Allemensen,
Ik heb gewoon alles wat ik heb kunnen wensen;
Wat is het fijn om gelukkig te zijn!
Contents
Chapter 1: Introduction: Intestinal interactions between the epithelial
barrier, the immune system and the nervous system
9
Part 1: Apoptosis and soluble tumour necrosis factor inhibitors in
inflammatory bowel diseases
45
Chapter 2: Apoptosis as a therapeutic paradigm in inflammatory bowel
diseases
47
Chapter 3: Inhibition of soluble TNF- by single domain camel antibodies
does not prevent experimental colitis
67
Part 2: Dendritic cell populations and C-type lectins in inflammatory
bowel diseases
87
Chapter 4: Dendritic cell populations in colon and mesenteric lymph nodes
of patients with Crohn’s disease
89
Chapter 5: Single nucleotide polymorphisms in C-type lectin genes,
clustered in the IBD2 and IBD6 susceptibility loci, may play a role in the
pathogenesis of inflammatory bowel diseases
109
Part 3: Cholinergic modulation of intestinal immune responses
127
Chapter 6: The enteric nervous system as regulator of intestinal epithelial
barrier function in health and disease
129
Chapter 7: Selective 7 nicotinic acetylcholine receptor agonists worsen
disease in experimental colitis
167
Chapter 8: Acetylcholine protects against inflammatory cytokine induced
epithelial barrier dysfunction
197
Chapter 9:
Summary and discussion
Nederlandse samenvatting
223
225
231
Chapter 10: Appendices
Dankwoord
Curriculum Vitae
List of publications
237
239
241
243
Chapter
1.
Introduction:
Intestinal interactions between the
epithelial barrier, the immune system and
the nervous system
Marleen I. Verstege1, Anje A. te Velde1, Wouter J. de Jonge1, Guy E. Boeckxstaens1,2
1. Tytgat Institute for Liver and Intestinal Research, Academic Medical Centre,
Amsterdam, The Netherlands
2. Department of Gastroenterology, Catholic University of Leuven, Leuven, Belgium
Chapter 1
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis
(UC), are chronic inflammatory diseases of the intestine of unknown aetiology. Although
the pathogenesis of these diseases is not well understood, several components of the
bacterial flora, the epithelial barrier, the immune system, the nervous system and mutations
in genes that are a part of these components have been shown to play an essential role in
mucosal inflammation. It has been demonstrated in animal models that the bacterial flora of
the intestines is involved in pathological processes of IBD; several mouse models that are
treated with antibiotics or that are housed in a germ-free environment do not develop colitis
1-5
. Moreover, IBD patients show increased mucosal secretion of IgG antibodies against
commensal bacteria of the resident flora
6,7
. A functional intestinal barrier is important to
prevent commensal bacteria to gain access to the lamina propria (LP) where they can
induce an inflammatory response.
We have investigated several aspects of the pathogenesis of IBD, which are
outlined in this thesis.
- 10 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
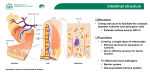
The epithelial barrier function
To prevent access of luminal contents to the LP, the epithelial layer has developed specific
barrier mechanisms, including adherens junctions, desmosomes, gap junctions and tight
junctions (TJs). TJs or zonula occludens are the most apical components of these
intercellular junctions. They prevent the diffusion of membrane proteins and lipids between
the basolateral and apical membranes so that cell polarity is preserved (fence function) and
a selective barrier to paracellular transport (barrier function) is formed. In contrast to
transcellular transport, which is highly selective because of ion channels and active
transport systems, paracellular transport is a rather passive process. It depends on ion and
molecular gradients at the basolateral and apical side and does not distinguish between
different ions and molecules. However, the barrier function of TJs restricts this paracellular
transport since TJs are selectively permeable for cations, water and small uncharged
molecules, whereas the passage of macromolecules is obstructed 8. Selectivity for ion size
and strength is different between tissues and is related to the composition of TJs.
TJ complexes are composed of a network of proteins that are coupled to actin
filaments of the cytoskeleton
8-10
. The proteins occludin (62-82 kDa), several members of
the claudin family (20-27 kDa) and junctional adhesion molecule (JAM) (36-41 kDa) make
up the membrane part of the TJ
11-14
. Although occludin and claudin demonstrate no
significant sequence similarity, they are both tetraspan proteins with two extracellular and
one intracellular loop and an intracellular N- and C-terminus. To integrate in the TJ, it is
essential that occludin is phosphorylated, whereas dephosphorylation redirects occludin to
intracellular
pools
decreasing
transepithelial
electrical
resistance
(TEER)
15-18
.
Nevertheless, occludin-deficient mice are still able to form well-developed TJ strands and
retain normal intestinal barrier integrity
19,20
. Consequently, occludin may increase the
strength of TJs, but other TJ proteins are obviously more essential. It seems that occludin is
more involved in cell signalling than in maintaining the epithelial barrier
21
. Claudins,
which consist of a family of at least 24 members, are probably the main barrier-forming
proteins. Since different types of claudins are expressed in a restricted number of cell types
or during periods of development, claudins are expected to contribute to tissue-specific
- 11 -
Chapter 1
functions of TJs. Intestinal epithelial cells express several claudins. It is assumed that
claudin-2 has the potential to form aqueous channels, whereas the permeability of
macromolecules is not increased 22. Overexpression of claudin-1 and -4 results in increased
TEER, indicating that these proteins are involved in tightening the paracellular barrier 23,24.
In CD patients it has been demonstrated that the pore-forming claudin-2 is upregulated and
that the sealing claudins 5 and 8 are downregulated
25
. JAM and occludin have been
implicated in the transmigration of leukocytes through the endothelial and epithelial
barriers, respectively
induced colitis
14,26
. Mice that are JAM deficient are more susceptible for DSS-
27,28
.
Members of the zonula occludens (ZO) family are proteins that form a bridge
between these membrane proteins and actin filaments, which are connected to the
perijunctional ring, a component of the cellular cytoskeleton
29-31
. ZO-1 proteins belong to
the membrane-associated guanylate kinase (MAGuK) homologue family, containing three
PDZ [postsynaptic density-95 (PSD-95)/Discs large (Dig)/ZO-1] domains, an SH3 domain
and a guanylate kinase (GuK) domain 32. These and some other domains are essential in the
bridge function of the ZO proteins. ZO-1 interacts with ZO-2 and -3 by PDZ domains. The
PDZ-1 domain is necessary to interact with the PDZ regions at the C-terminus of claudins
29-31
. The GuK region of ZO-1 mediates binding to the C-terminus of occludin
29-31,33
.
Besides, the SH3 region of ZO-1 mediates binding to G proteins, like Gi2, and the Cterminus of ZO proteins interacts to F-actin 29,31,34. The function of ZO-1 is not exclusively
restricted to the organisation of TJs, as it is also detected in the nucleus where it regulates
cell growth and differentiation 35,36. The expression of ZO-1 in colonic epithelium is lost in
DSS-induced colitis in mice
37
. Also in colonic tissues of UC patients, the expression of
occludin, ZO-1, JAM and claudin-1 is downregulated 38.
Gi2 proteins are localised within the TJs and have an important function in the
maintenance and development of TJs, probably through the protein kinase C (PKC)
pathway that regulates the phosphorylation of the myosin light chain (MLC) 39-41. Mice that
are Gi2 deficient spontaneously develop colitis similar to that of human patients with UC,
clinically manifested by diarrhoea and bloody stools
- 12 -
42-46
. Phosphorylation of MLC causes
Intestinal interactions between the epithelial barrier, immune system and nervous system
contraction of the perijuctional ring, which is a component of the cellular cytoskeleton, so
that the permeability of TJs is increased
47-49
. MLC is phoshorylated by myosin light chain
kinase (MLCK), which is regulated by PKC. The activation of PKC is in turn regulated by
seven-membrane-helix receptors that are coupled to G proteins. G proteins are activated
following the binding of a ligand to its receptor. Thereafter the subunit of the G protein
activates phospholipase C (PLC)- that upregulates the second messengers diacylglycerol
(DAG) and IP3, so that Ca2+ is released from the endoplasmatic reticulum to the cytoplasm.
Ca2+ and DAG activate PKC and consequently MLCK is phosphorylated so that its activity
decreases MLC phosphorylation. In conclusion, activation of PKC proceeds to a decrease
in transcellular permeability and an increase in TEER.
Enteric pathogens have developed several mechanisms to disrupt TJs of epithelial
cells. This occurs mainly by modulating the perijunctional actomyosin ring or by interfering
with TJ proteins directly
50-53
. Bacterial products degenerate or (de)phosphorylate specific
TJ proteins or use them as a receptor so that these proteins become dysfunctional resulting
in a decrease of the efficacy of the TJ. The latter is manifested as a decrease of TEER and
as an increase of the paracellular flux of macromolecules like mannitol, often clinically
resulting in diarrhoea.
In IBD the epithelial layer is inflamed without obvious exogenous factors like a
(bacterial) infection. Nevertheless, colonic biopsies from CD patients contain decreased
numbers of Lactobaccillus and Bifidobacteria, whereas the mucosa and probably even the
intraepithelial layer contain an increased population of adherent bacteria
54-56
. Increasing
evidence suggests that the immune system itself modulates TJs and intestinal permeability.
IBD patients have increased concentrations of pro-inflammatory cytokines, like tumour
necrosis factor- (TNF-), interferon- (IFN-), interleukin (IL)-8 and IL-1
. In vitro
57-59
studies have demonstrated that these cytokines decrease the barrier function of intestinal
epithelial monolayers and induce reorganisation of several TJ-associated proteins, including
ZO-1, JAM-1, occludin and claudin-1, and -4
60-64
. An increase of the intestinal
permeability caused by an activated IFN- receptor complex is also associated with a
- 13 -
Chapter 1
decrease in the PLC- activity resulting in MLC phosphorylation
64
. It seems that IL-1
increases intestinal permeability by the induction of MLCK gene transcription and
consequently increases MLCK protein activity, probably mediated by a rapid activation of
the transcription factor nuclear factor (NF)-B
57,65
. IL-1-mediated increased intestinal
permeability leads to an increased paracellular transport of luminal antigens 65-68 Also TNFmediated increased intestinal permeability seems to be NF-B dependent and leads to a
downregulation of ZO-1 proteins and alteration in junctional localisation
69
. In IBD, the
intestinal permeability could be increased because of the effects on the epithelial barrier by
these pro-inflammatory cytokines, which are increased in IBD patients. In chapter 8 we
demonstrate that the neurotransmitter acetylcholine (Ach) and muscarine decrease NF-B
activation and decrease IL-1- and TNF--induced production of IL-8 by epithelial cells.
Moreover, Ach and muscarine protect against cytokine-induced enhanced permeability.
- 14 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
Antigen interaction with dendritic cells
Specialised epithelial cells termed M (microfold) cells, which are scattered among the
epithelial cells (ECs) in the follicle-associated epithelium above the Peyer’s patches (PPs)
are able to absorb, transport, process and present (microbial) antigens to dendritic cells
(DCs) in the subepithelial dome (SED) of the PP. In humans, CD11c+ DCs are concentrated
in the SED and T cell zones and are particularly involved in the activation of T cells that
support IgA-class switching by B cells and in the induction of oral tolerance. Besides, DCs
are located in the LP just below the basement membrane, where they interact with antigens
that have gained access to the LP following disruption of the epithelial barrier because of
infection and/or inflammation, as presumably appears in IBD. In addition, DCs sample
antigen directly by expanding dendrites among ECs into the lumen. These DCs are capable
to open TJs between enterocytes, since they modulate different TJ proteins, including JAM1, claudin-1 and ZO-1
70
. The chemokine receptor fractalkine (CX3CR1) on LP DCs
enables them to sample luminal antigens directly by transepithelial dendrite interaction with
epithelial CX3CL1
71
. The authors suggest that the interaction between CX3CR1 and
CX3CL1 is responsible for the accumulation of DCs and T cells in the LP of IBD patients.
Rimoldi et al. have shown that the intestinal homeostasis is regulated by the interaction
between ECs and DCs: EC-conditioned DCs produce IL-10 and IL-6, but not IL-12 and
induce a Th2 response, even in the presence of pathogens that normally promote a Th1
response
72
. It is possible that CD patients lack an adequate interaction between ECs and
DCs resulting in a Th1-mediated inflammation. Moreover antigens are taken up by DCs
indirectly by internalising apoptotic ECs and by taking up antigen-containing exosomes
shed from ECs
73
. Exosomes are small membrane-bound vesicles, which are not only
secreted by ECs, but also by haematopoietic cells, including DCs and other certain cell
types. It has been shown that immunosuppressive DC-derived exosomes are capable to
suppress inflammatory responses in rheumatic arthritis. The exact mechanism is not clear,
but it is likely that DC-derived exosomes are internalised by endogenous or follicular DCs
to transfer molecules like MHC class II molecules so that antigen-specific T cell responses
are induced.
- 15 -
Chapter 1
Dendritic cell populations
Human DCs express high levels of human leukocyte antigen (HLA)-DR and are lineage
negative, which indicate that they do not express specific markers of B and T cells,
monocytes and natural killer cells. In peripheral blood five distinct subsets of DCs can be
distinguished, namely CD1c+ (BDCA1), CD16+, BDCA3+, CD123+ and CD34+ DCs
74
.
Myeloid precursor DCs express high levels of CD11c and can be distinguished in DCs that
express CD1b/CD1c, CD16 or BDCA3
75
. These DC populations produce IL-12 in
response to bacterial compounds or CD40L and are GM-CSF-dependent for survival. In
contrast, plasmacytoid DCs are CD11c negative and express CD123, BDCA2 and BDCA4
76,77
. These IL-3 dependent DCs respond especially in case of viral infections by the
production of type I interferon (IFN), including IFN- and IFN-.
In the gut, DCs are present in the primary sites for the induction of intestinal T and
B cell responses which include the PPs located in the small intestine, isolated lymphoid
follicles in the colon and mesenteric lymph nodes (MLNs), all structures that are part of the
gut-associated lymphoid tissue (GALT). Te Velde et al. had demonstrated two distinct DC
subpopulations in IBD patients: an ICAM-3 grabbing non-integrin (DC-SIGN)+ population
that was present scattered throughout the mucosa and a CD83+ population that was present
in aggregated lymphoid nodules and as single cells in the LP
78
. Only DC-SIGN+ DCs
produce the pro-inflammatory cytokines IL-12 and IL-18. Interestingly, in CD patients the
expression of both populations was increased compared to healthy controls. In chapter 4
we demonstrate that the myeloid DC populations positive for CD1a and BDCA-1 are absent
in colonic mucosa and MLNs, whereas BDCA3+ DCs are highly expressed throughout the
LP and around (sub)capsular and medullary sinuses, blood vessels and B-cell follicles in
the MLN
79
. In MLNs and lymph follicles in the colon the expression of s-100+ DCs is
increased in CD patients.
- 16 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
Antigen recognition by DCs
Since the gut contains massive numbers of microbes, it necessary that the immune system is
able to discriminate between commensal and pathogenic microbes. Therefore DCs and
other immune cells such as macrophages are able to recognise pathogen-associated
molecular patterns (PAMPs) through binding to pattern recognition receptors (PRRs) on
their membrane 80. Most important PRRs are the Toll-like receptors (TLRs), the nucleotide
oligomerisation domain (NOD)-like receptors (NLRs) and the C-type lectins.
Toll-like receptors
TLRs are highly conserved proteins and so far, eleven members of the TLR family have
been identified in mammals. Toll was first identified as a protein involved in the controlled
dorsoventral formation during the (embryonic) development of the Drosophila
melanogaster. Drosophila that are Toll deficient are not able to clear infections caused by
fungi, since some antimicrobial products will not be produced. Because Drosophila does
not have an adaptive immune system, TLRs are involved in an evolutionary conserved
signal pathway that induces innate immune responses. TLRs are characterised by aminoterminal leucine-rich repeats that are responsible for the recognition of PAMPs and they
possess a carboxy-terminal Toll-IL-1 receptor (TIR) domain of which the sequence is
homologous to that of interleukin receptor-1 (IL-R1) family proteins. Each TLR recognises
different PAMPs and the first human TLR member to be discovered was TLR4, which is a
transmembrane protein that has to be demonstrated the receptor for lipopolysaccharide
(LPS), a component of the outer membrane of gram-negative bacteria
81-83
. Recognition of
LPS by TLR4 is a complex process and several accessory molecules are necessary. First
LPS has to bind to the plasma protein LBP (LPS-binding protein) so that it is able to
interact with the soluble or GPI-anchored protein CD14, which is produced by monocytederived cells and this complex binds to TLR4
are hyporesponsiveness to LPS
84,85
. Mice that are TLR4 or CD14 deficient
81,86
. Individuals with a mutation in TLR4 have a slightly
87
increased risk to develop CD .
- 17 -
Chapter 1
TLR2 recognises different components of mainly bacteria and yeast, such as
peptidoglycan from gram-positive bacteria, lipoteichoid acid, zymosan from yeast and
lipoproteins. TLR2 forms heterodimers with TLR1 when activated by bacterial
lipoproteins, whereas mycoplasma-derived lipoprotein triggers TLR2 to form heterodimers
with TLR6 88,89.
TLR5 recognises flagellin, which is a protein that forms bacterial flagella
90
.
Intestinal ECs express TLR5 at the basolateral side where they can sense flagellin from
pathogenic bacteria such as Salmonella
91
. Mice lacking TLR5 develop colitis
92
spontaneously . Interestingly mice that are both TLR5 and TLR4 deficient have elevated
bacterial loads in the colon; however they do not develop colitis 92. Serum IgG to flagellin
is elevated in CD and UC patients and a dominant-negative TLR5 polymorphism reduces
adaptive immune responses to flagellin and in some ethnicities heterozygous carriage is
associated with a protection from CD 93,94.
TLR3, TLR7, TLR8 and TLR9 are intracellular receptors present in endosomal
compartments and are specialised in the recognition of nucleic acids. TLR3 recognises
double stranded (ds)RNA generated during the replication of viruses. It has been shown
that mice that are TLR3 deficient are more susceptible for infections with cytomegalovirus
and West Nile virus 95. DSS-induced colitis in mice is ameliorated by systemic, but not oral
administration of synthetic viral RNA that activates TLR3 95. TLR7 and TLR8 are involved
in the recognition of single stranded (ss)RNA rich in guanosine or uridine derived from
RNA viruses
96
. Mice that are TLR7 deficient are not capable to induce inflammatory
cytokines, type I IFN and plasmacytoid DC maturation
97
. Although TLR7 and TLR8
recognise both ssRNA, TLR7 activation is characterised by a strong induction of type I
IFNs, whereas TLR8 activation results in the induction of pro-inflammatory cytokines as
TNF-
98,99
. Unmethylated 2'-deoxyribo (cytidine-phosphate-guanosine) (CpG) DNA
motifs found in bacteria and several viruses are recognised by TLR9. TLR9-dependent
activation by DNA-containing immune complexes seems to be mediated by the highmobility group box-1 protein (HMGB-1) and the receptor for advanced glycosylated end
products (RAGE)
- 18 -
100,101
. DSS-induced colitis is exacerbated in TLR9-deficient mice,
Intestinal interactions between the epithelial barrier, immune system and nervous system
probably because of a disturbed homeostasis 102. It has been shown that TLR9 at the apical
site of the epithelial barrier does not give an immune reaction as a result of binding its
ligand CpG DNA
102-104
. However, micro-organisms that pass the epithelial barrier are
recognised by TLR9 at the basolateral site resulting in activation of the NF-B pathway.
Most TLRs activate the transcription factor NF-B through a myeloid
differentiation factor 88 (MyD88)-dependent pathway, resulting in the expression of genes
encoding for inflammatory cytokines, including TNF-, IL-6 and IL-1. All TLRs, with the
exception of TLR3, recruit the intracellular protein MyD88 through TIR domain
interactions. These interactions result in the recruitment of IL-R1 associated kinase
(IRAK)-1 and -4 to arrange a complex
susceptible for DSS-induced colitis
105,106
. Mice that are MyD88 deficient are more
107
. Recognition of commensal bacteria seems to be
important for maintaining the integrity of the epithelial barrier. Macrophages of MyD88deficient mice are not able to activate IRAK-1 after exposure of LPS and the production of
TNF-, IL-6 and IL-1 is inhibited 108. IRAK-1 is a serine-threonin kinase of which the Nterminal region contains a death domain that interacts with the death domain of MyD88 109.
Mice that are deficient for IRAK-1 confirm an insufficient response to LPS 110. The adaptor
protein TNF receptor associated factor 6 (TRAF-6) is also recruited to the complex by
association to phosphorylated IRAK-1. TRAF-6-deficient mice have osteoporosis and
macrophages derived from the bone marrow of these animals are insufficient in the
production of nitric oxide in response to LPS
111
. Phosphorylated IRAK-1 and TRAF-6
dissociate from this complex to form a complex with transforming growthfactor activated
kinase (TAK)-1, TAK-1 binding protein (TAB1) and TAB2 at the plasmamembrane
resulting in the phosphorylation of TAB2 and TAK1. IRAK-1 is degraded and
ubiquitylation of TRAF-6 leads to the activation of TAK1, which phosphorylates both
mitogen-activated protein (MAP) kinases and the inhibitor of nuclear factor IB kinase
(IKK) complex. The IKK complex consists of two catalytic subunits, IKK and IKK and
one regulatory subunit, IKK or NF-B essential modulator (NEMO). Activation of this
complex results in the phosphorylation of IBs so that NF-B translocates from the cytosol
into the nucleus where it induces the expression of its target genes. Although NF-B is
- 19 -
Chapter 1
thought to induce the transcription of mainly pro-inflammatory genes, mice that are NEMO
deficient and consequently do not signal via the NF-B pathway develop colitis
spontaneously
112
. This indicates that NF-B signalling regulates epithelial integrity and
intestinal immune homeostasis.
Nucleotide oligomerisation domain-like receptors
NLRs are intracellular recognition proteins that contain similar to TLRs C-terminal leucinerich repeats and an N-terminus consisting of protein-protein interaction domains, such as
caspase recruitment domains (CARD) or pyrin domains. NOD1 is involved in the
recognition of -D-glutamyl-meso-diaminopimelic acid (ie-DAP), which is a cell-wall
derivate of gram-negative bacteria, whereas muramyl dipeptide (MDP), a component of
both gram-negative and -positive bacterial peptidoglycan (PGN), is a ligand for NOD2
113,114
. In theory, mutations in NOD2 will result in a decreased activation of the NF-B
pathway and consequently in a decreased production of pro-inflammatory cytokines.
However, mouse models and family studies have revealed that mutations in NOD2 are
associated with the development of CD accompanied with an increased production of proinflammatory cytokines including TNF- and IL-12 87,115-117. It is still not clear what causes
this discrepancy and whether a mutation in NOD2 will result in a gain
function
115,117
116
or a loss of
. Different mutations in NOD2 have been described in which the
substitutions R702W and G908R and the C-insertion mutation at nucleotide 3020
(3020inC) are most common in humans 118,119. Mutations associated with CD are located in
the leucine-rich repeats, whereas mutations in the NACHT domain results in Blau
syndrome 120.
MDP is not only recognised by NOD2, but also another member of the NLR
family, NALP3/CIAS1/cryopyrin/NLRP3, is activated by MDP
121
. NALP3 has a similar
structure of NOD2, but contains a pyrin domain instead of a CARD domain. Gain-offunctions mutations in the NACHT domain of NALP3 are associated with three autoinflammatory diseases, Muckle-wells syndrome (MWS), familial cold auto-inflammatory
syndrome (FCAS) and chronic infantile neurological cutaneous and artricular syndrome
- 20 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
(CINCA), which are characterised by periodic fever syndromes
122
. Recently is discovered
that a polymorphism in NALP3 is also associated with an increased risk of CD 123. NALP3
is involved in the activation of the pro-inflammatory cytokines IL-1 and IL-18 through the
activation of caspase-1 that cleaves the inactive cytoplasmic precursors pro-IL-1 and proIL18 into its mature active forms
124
. Activated NALP3 forms together with two adapter
molecules ASC and CARDINAL the so-called ‘inflammasone’ resulting in the recruitment
of two caspase-1 molecules and consequently in the induction of active IL-1 and IL-18 125.
These cytokines seem to be important in the pathogenesis of CD since IL-1 production is
increased in morphological normal intestinal biopsies from patients with CD 126 and in mice
it has been shown that neutralisation of IL-18 ameliorates TNBS-induced colitis 127.
C-type lectins
C-type lectins are transmembrane proteins that recognise carbohydrate structures in a
calcium-dependent manner using highly conserved carbohydrate recognition domains
(CRDs)
128
. Glycosylated molecules and micro-organisms that bind to C-type lectins
expressed by DCs will be internalised, processed in endosomes and finally presented to T
cells, together with MHC class I and II molecules. Since in the cytoplasmic regions of Ctype lectins immunoreceptor tyrosine based activation (or inhibitory) motifs (ITAMs or
ITIMs) are present, activation of C-type lectins may result in pro- or anti-inflammatory
responses
129,130
. In contrast to TLRs and NOD proteins, C-type lectins recognise not only
foreign, but also self-proteins, so that activation of C-type lectins will not always result in
DC maturation and T cell activation
131,132
. The balance between the activation of C-type
lectins and TLRs regulates the outcome of the innate immune response 133.
Several micro-organisms such as HIV, dengue virus, hepatitis C virus,
Mycobaterium tuberculosis and Candida albicans interact with the C-type lectin DC-SIGN
134-138
. Nevertheless, activation of DC-SIGN alone will not result in sufficient innate
immune responses; however DC-SIGN ligation on DCs after TLR4 activation increases
cytokine production dramatically
139,140
. Nagaoka et al. demonstrated that DC-SIGN
associates with the TLR4-MD-2 complex and that signal transduction by the recognition of
- 21 -
Chapter 1
LPS in gram-negative bacteria is enhanced 141. Interaction of the C-type lectin dectin-1 with
TLR2 results in the generation of pro-inflammatory responses to fungal pathogens 142,143.
In CD patients, the expression of a subpopulation DC-SIGN+IL-12+IL18+ DCs is
increased in colonic mucosa compared to healthy controls 78. In Chapter 5 we demonstrate
that polymorphisms in the C-type lectins DC-SIGN, dendritic cell immuno receptor (DCIR)
and macrophage galactose-like lectin (MGL) are not associated with IBD. However,
polymorphisms in lectin-like transcript 1 (LLT1) seems to be associated with a slightly
increased risk of CD.
- 22 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
Antigen presentation by DCs
Upon antigen encounter, immature DCs are activated and undergo a differentiation process
in which they fully mature into highly stimulatory antigen presenting cells. During this
process they lose their endocytotic capacity that was necessary for the uptake and
processing of antigens in the periphery. In addition, they upregulate the chemokine receptor
CCR7 so that they are able to migrate to the draining lymph node where they encounter
naïve populations of T cells. Moreover, mature DCs upregulate co-stimulatory molecules,
including CD80 (B7.1), CD86 (B7.2) and CD40 and adhesion molecules such as ICAM-1
and LFA-1 144. In the lymph node, mature DCs present the processed antigen in association
with MHC class I and II molecules to naïve T cells so that these T cells become activated
and differentiate into effector T cells. Naïve T cells that recognise the antigen, but that are
not co-stimulated become anergic in which T cells become unresponsiveness. Depending
on which PRRs are activated by the captured antigens, DCs will direct naïve T cells to
differentiate into T helper (Th) 1, Th2, Th17 or regulatory T cells. In conclusion, to activate
T cells, three signals of DCs are necessary: 1) a peptide/MHC complex that is recognised
by the T cell receptor, 2) sufficient co-stimulation to prevent anergy and 3) T cell
polarisation signals to adapt the effector phenotype.
Depending on the interaction between DCs and different micro-organisms, DCs
produce high or low concentrations of IL-12, which is an important determinant of the
direction of the immune response. High concentrations of IL-12 will direct T cells to
develop into Th1 cells, whereas low concentrations allow for the production of IL-4 by the
T cell pool itself which, in turn, will accelerate the development of Th2 cells. IL-10
production by (regulatory) DCs may facilitate the generation of regulatory T cells, which
are important in the induction of tolerance to self and harmless foreign antigens.
It has been demonstrated that an exaggerated immune response against the
endogenous microflora by Th1 and Th17 lymphocytes plays an important role in the
pathogenesis of CD. This immune response is characterised by an increase of proinflammatory cytokines, including TNF-, IL-1, transforming growth factor (TGF)-
- 23 -
Chapter 1
IFN- and IL-17 in the inflamed mucosa of CD patients. High concentrations of TNF-
can also be detected in the stool of CD patients
145,146
. Overexpression of TNF- in mice
results in the development of chronic inflammatory arthritis and Crohn’s like IBD
147
.
TNF- is present in two forms, namely as a transmembrane and as a soluble protein. Since
TNF- seems to be a key player in the pathogenesis of CD, pharmaceutical industries
developed TNF- inhibitors. However, these drugs have side-effects which include
immunoreactivity. In chapter 3 we investigated TNF- inhibitors based on the light chains
of camel antibodies in an acute and a chronic colitis model. Unfortunately, these TNF-
inhibitors did not ameliorate colitis, probably since only the soluble form of TNF- is
blocked and not the transmembrane form. Blocking of only soluble TNF- may lead to an
impaired apoptosis in IBD patients resulting in survival of reactive T cells which can
maintenance inflammatory processes. Moreover, the transmembrane form of TNF- seems
to be more involved in cell survival processes instead of cell death 148,149. In chapter 2 we
discuss apoptotic mechanisms and their association to IBD. In addition, we will review how
specific therapeutic approaches interact at different levels with the apoptotic pathway.
Tolerance and regulatory T cells
Tolerance is achieved by different mechanisms, both thymic and peripheral, to prevent
accumulation and activation of auto-reactive T cells. In the thymus auto-reactive T cells are
eliminated by negative selection, i.e. T cells with specificity for self antigens become
apoptotic and are deleted 150. Nevertheless, a population of low-affinity self-reactive T cells
will escape this thymic selection process and enter the circulation to the periphery. Here
peripheral tolerance mechanisms take over to prevent auto-reactivity. Auto-reactive T cells
are inhibited by regulatory (suppressor) T cells, a diverse subset of CD4+ T cells, including
CD4+CD25+ cells, Tr1 and Th3 cells 151,152. Failure of one of these mechanisms may result
in allergies, rejection of transplanted organs or autoimmune diseases, such as type I
diabetes and multiple sclerosis, but also IBD is probably caused by impaired regulatory
mechanisms, so that overwhelming Th1 responses can be developed.
- 24 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
Regulatory T cells can be distinguished in naturally occurring regulatory T cells
and adaptive regulatory T cells
153
. Naturally occurring T cells acquire their regulatory
function in the thymus during early neonatal development and migrate into peripheral tissue
were they suppress the proliferation and cytokine production of self-reactive T cells in a
mainly contact dependent manner to maintain tolerance to especially auto-antigens
+
151
Since they express high levels of the IL-2 receptor (CD25) they are referred as CD4 CD25
.
+
cells, although also activated CD4+ T cells express CD25. Moreover they are characterised
by high membrane expression of CD38, CD62L, CD103 and glucocorticoid-induced TNF
receptor (GITR), cytotoxic T lymphocyte antigen 4 (CTLA-4 or CD152) and by the
expression of FoxP3.
On the contrary, adaptive regulatory T cells, including Tr1 and Th3, are dependent
on antigen presentation of DCs and are mainly involved in the mucosal tolerance to
widespread antigens and commensal microflora, predominantly by the production of antiinflammatory cytokines, including IL-10 and TGF- 154,155. Th3 cells produce high levels of
TGF- and were first identified in oral tolerance studies
high levels of IL-10
156,157
, whereas Tr1 cells produce
158
. Immature DCs were shown to induce the development of Tr1 cells
through the production of TGF- and IL-10, both in vitro 159 and in vivo 160. However, also
161
mature DCs can induce regulatory T cells
, dependent on culture conditions and the
priming antigen.
Immature regulatory DCs can be induced by the hepatitis C virus and
Mycobacterium tuberculosis. In contrast, schistosoma-derived lysophosphatidylserine,
filamentous haemagglutinin of Bordetella pertussis, cholera toxin B and fungus-derived
cordycepin induce mature regulatory DCs that produce variable amounts of IL-10, but all
induce Tr1 cells
. Filamentous haemagglutinin of Bordetella pertusssis has been
162-165
shown to ameliorate the disease activity in a chronic T cell dependent colitis model by the
induction of anti-inflammatory cytokines
166
. Furthermore also commensal microbes such
as lactobacilli, which act through DC-SIGN and Mycobacterium vaccae have been
associated with the induction of mature regulatory DCs and the generation of regulatory T
cells 161,167,168. Characteristically, mainly pathogens that induce chronic diseases are able to
- 25 -
Chapter 1
suppress the immune response by the activation of (IL-10-producing) regulatory DCs,
either immature or mature.
Since several commensal bacteria (i.e. probiotics) and helminths like Trichuiris
suis decrease the production of the Th1-associated cytokine IL-12 by DCs and increase the
production of IL-10, resulting in an inhibition of the generation of Th1 cells
169,170
, they
could be a potential treatment of IBD. Oral intake of genetically modified probiotic
Lactococcus lactis that produce IL-10 has been shown to decrease disease activity in CD
patients in a phase I clinical trial
171
. It is likely that in IBD patients the balance between
regulatory T cells and Th1 cells is disturbed, so that mainly Th1 responses are activated.
Probably by the generation of regulatory DCs, the fate of T cells can be changed in T cells
that gain a regulatory function instead of T cells that induce inflammation. When we know
more about responses of DCs and the fate of T cells reacting to probiotica, commensal
bacteria and pathogens, we will understand more how DCs discriminate between different
micro-organisms. This could be a rationale for DC immunotherapy, which is also one of the
therapeutic approaches in other autoimmune diseases, such as diabetes and multiple
sclerosis, cancer and allergies.
Th17 cells
A novel T helper cell lineage, Th17 that exclusively produces the pro-inflammatory
cytokine Th17 has been reported to play an important role in many inflammatory diseases,
including IBD. The IL-12 family member IL-23 is produced by DCs and promotes the
differentiation of CD4+ T cells that produce IL-17 and seems to play an important role in
regulating the Th1/Th17 balance in IBD
172-174
. A genome-wide association study showed
that a polymorphism in the receptor for IL-23 confers strong protection against CD
175
.
Moreover, anti-IL-23 therapy was effective in the prevention as well as the treatment of
active experimental colitis
176
. IL-17 expression and Th17 differentiation is downregulated
by IFN- in experimental colitis and UC patients that receive IFN- therapy
177
. Besides
the production of IL-17, Th17 cells produce other pro-inflammatory cytokines including
IL-21, IL-22, TNF- and IL-6 174,178. IL17 and IL-21 are overexpressed in colonic samples
- 26 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
from IBD patients and neutralisation of IL-21 reduces the secretion of IL-17 by LP T
lymphocytes derived from CD patients 179. Moreover, both TNBS- and DSS-induced colitis
are ameliorated in IL-21-deficient mice, probably since naïve T cells from these mice failed
to differentiate into Th17 cells 179. In experimental colitis, IL-21 prevents TGF--dependent
expression of FoxP3 resulting in a reduction of regulatory T cells
180
. TGF-1 is able to
differentiate naïve T cells into regulatory T cells, which prevent autoimmunity
180
.
However, in the presence of IL-6, TGF-1 has been shown to converts naïve T cells into
Th17 cells
180
. It seems that the vitamin A metabolite retinoic acid plays an important role
in the regulation of TGF-1-dependent immune responses in which retinoic acid inhibits
the IL-6- and IL-23-driven induction of Th17 cells and promotes FoxP3+ regulatory T cells
differentiation by enhancing TGF--driven Smad-3 signalling 181,182. In chapter 4 we show
that polymorphisms in LLT1 are slightly associated with CD. Interestingly, LLT1 is a
ligand for CD161, which is a new surface marker for human IL-17 producing Th17 cells
183,184
. It has been shown that CD161+CD4+ T cells are a resting Th17 pool that can be
activated by IL-23 and mediate destructive tissue inflammation in the intestines of CD
patients 184.
- 27 -
Chapter 1
The cholinergic pathway in immune regulation and intestinal epithelial
barrier function
Besides the intestinal epithelial barrier function and the intestinal immune system, the
nervous system plays an important role in the homeostasis of the gut. The intestinal tract is
innervated by the vagus nerve, which is part of the parasympathetic nervous system known
to regulate heart rate, hormone secretion, gut motility, respiratory rate, blood pressure and
other vital processes of the body. The two vagus nerves originate in the medulla oblongata
and preganglionic fibres travel uninterrupted to the organs they innervate. There the
preganglionic fibres synapse with short postsynaptic fibres that are distributed throughout
the organ. Ach is the principal neurotransmitter of the vagus nerve and plays a key role in
the anti-inflammatory pathway.
The enteric nervous system (ENS) is an integrated network of neurons and enteric
glial cells (EGCs) and is organised in a submucosal plexus or Meissner’s plexus located
between the mucosa and the circular muscle layer, and a myenteric plexus or Auerbach’s
plexus located between the circular and longitudinal muscle layers. The ENS is regulated
by the central nervous system, but is in contrast to other organs also able to function
independently. In general, the Meissner’s plexus regulates secretory responses of the
mucosa, whereas the Auerbach’s plexus is involved in the regulation of gastrointestinal
motility. It has been shown that besides neurones also EGCs modulate gastrointestinal
functions indirect or directly 185,186.
Interactions between the nervous system and the immune system
It has been described that cholinergic activation has anti-inflammatory effects in several
diseases
187-193
. Vagotomy and cholinergic antagonists have been shown to worsen
inflammation in animal colitis models, whereas stimulation of the vagus nerve results in an
amelioration of postoperative ileus in part through its anti-inflammatory effects
187,188,194
.
Most effects of the vagus nerve have been based on the effects of Ach, which signals
through either muscarinic receptors or nicotinic receptors. Macrophages and also other
- 28 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
immune cells like DCs express several subunits of the nicotinic acetylcholine receptors
(nAchRs) such as the 4, 2 and 7 subunit
. Selective 7 nAchR agonists
195,196,own data
have been shown to ameliorate pancreatitis, DSS-induced colitis and postoperative ileus
188,191,193,194
. Nicotine, which acts through the 7 homopentamer, inhibits the production of
pro-inflammatory cytokines and chemokines in macrophages and inhibits the NF-B
pathway and HMGB1 secretion
187,197
. Interestingly, nicotine has also beneficial effects in
several subgroups of patients with UC, but not in CD patients
194,198,199
. Also Ach itself
inhibits the release of pro-inflammatory cytokines such as TNF-, IL-1, IL-6 and IL-18
by macrophages, but stimulated with endotoxin the production of the anti-inflammatory
cytokine IL-10 is not affected
. In chapter 7 we tested two new selective 7 nAchR
195
agonists in two different mouse models. Although earlier research demonstrated that
activation of the vagus nerve ameliorate intestinal inflammation, we show that both 7
nAchR agonists worsen colitis or are ineffective.
Besides Ach also other neuropeptides have been implicated to be antiinflammatory. Cholecystokinin (CCK) is responsible for the activation of digestion of
dietary fat and it is indicated that CCK reduces TNF- and IL-6 release in haemorrhagic
shock by the intake of high-fat nutrition
189
. Vagotomy abrogates this anti-inflammatory
effect of both high-fat intake and CKK, indicating that the vagus nerve is responsible for
CCK-reduced inflammation. Vasoactive intestinal peptide (VIP) regulates the secretion of
water and electrolytes and the dilation of the smooth muscles of the gut to increase gut
motility. In TNBS-induced colitis, VIP ameliorates clinical symptoms and microscopic
inflammation by regulating the balance between Th1, Th2 and Th17 differentiation 200.
Interactions between the nervous system and the epithelial barrier
Although in general activation of the cholinergic anti-inflammatory pathway and the release
of VIP and Ach leads to decreased inflammation in several diseases, it also results in an
increase of intestinal permeability 201. Both VIP and Ach increase paracellular permeability
in the gut
202,203
. Moreover, it has been demonstrated that the release of VIP inhibits
- 29 -
Chapter 1
proliferation of epithelial cells and that it is necessary to maintain intestinal epithelial
barrier integrity
203,204
. Besides paracellular transport, Ach is also able to increase
transcellular transport via muscarinic Ach receptor activation
205
. These results seem to be
contradictory since an increased intestinal epithelial barrier leads to an increased influx of
antigens into the intestinal mucosa where they can induce an immune reaction.
In contrast to the cholinergic pathway, EGCs seem to decrease intestinal
permeability since ablation of EGCs in transgenic mice causes an increase of intestinal
permeability and causes intestinal inflammation 206. Furthermore, in vitro co-culture models
of EGCs and intestinal epithelial cell lines demonstrate that EGCs decrease the
permeability, probably via the release of S-nitrosoglutathione and the regulation of ZO-1
and occludin expression 206. S-nitrosoglutathione is able to restore mucosal barrier function
in colonic biopsies from CD patients 206. Moreover, EGCs inhibit proliferation of intestinal
epithelial cells which is partly TGF-1 dependent
207
. Mice that are deficient for EGCs
show an increased uptake of thymidine in intestinal ECs and crypt hyperplasia 207. Probably
an interaction between enteric neurones and EGCs is necessary to maintain epithelial
barrier function homeostasis, since in general the cholinergic pathway increases intestinal
permeability, whereas EGCs do the opposite. In chapter 6 we give an overview of how
neurotransmitters influences epithelial barrier function. In chapter 8 we show that
intestinal permeability is mainly decreased through the activation of muscarinic receptors
under inflamed conditions.
- 30 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
Thesis outline
Since many components are involved in the pathogenesis of IBD, it is important to
understand how the environment (gut flora and food antigens), epithelial barrier, immune
system, nervous system and genetic make-up interact with each other. In this thesis we have
investigated different parts of the pathogenesis of IBD. The first part of this thesis describes
how apoptosis plays a role IBD (Chapter 2) and how we investigated a new TNF-
inhibitor in two different colitis models (Chapter 3). In the second part of this thesis we
investigated which DC populations are present in the colon and MLNs of CD patients
(Chapter 4) and whether mutations in genes that encodes several C-type lectins are
associated with IBD (Chapter 5). The last section of this thesis describes how the ENS
influences barrier function of the intestine (Chapter 6 and 8) and how we investigated two
new 7 nAchR agonists in two different experimental mouse models (Chapter 7).
- 31 -
Chapter 1
References
1 Hoentjen,F. et al. (2003) Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic
activities in various regions of the colon in interleukin 10 gene deficient mice. Gut 52, 1721-1727
2 Dianda,L. et al. (1997) T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a
microbial environment. Am. J. Pathol. 150, 91-97
3 Schultz,M. et al. (1999) IL-2-deficient mice raised under germfree conditions develop delayed mild focal
intestinal inflammation. Am. J. Physiol 276, G1461-G1472
4 Sellon,R.K. et al. (1998) Resident enteric bacteria are necessary for development of spontaneous colitis and
immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224-5231
5 Veltkamp,C. et al. (2001) Continuous stimulation by normal luminal bacteria is essential for the development
and perpetuation of colitis in Tg(epsilon26) mice. Gastroenterology 120, 900-913
6 Macpherson,A. et al. (1996) Mucosal antibodies in inflammatory bowel disease are directed against intestinal
bacteria. Gut 38, 365-375
7 Duchmann,R. et al. (1999) T cell specificity and cross reactivity towards enterobacteria, bacteroides,
bifidobacterium, and antigens from resident intestinal flora in humans. Gut 44, 812-818
8 Tsukita,S. et al. (2001) Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2, 285-293
9 Madara,J.L. (1987) Intestinal absorptive cell tight junctions are linked to cytoskeleton. Am. J. Physiol 253,
C171-C175
10 Nusrat,A. et al. (2000) Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight
junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am. J. Physiol Gastrointest. Liver
Physiol 279, G851-G857
11 Furuse,M. et al. (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell Biol.
123, 1777-1788
12 Furuse,M. et al. (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no
sequence similarity to occludin. J. Cell Biol. 141, 1539-1550
13 Heiskala,M. et al. (2001) The roles of claudin superfamily proteins in paracellular transport. Traffic. 2, 93-98
14 Martin-Padura,I. et al. (1998) Junctional adhesion molecule, a novel member of the immunoglobulin
superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142,
117-127
15 Andreeva,A.Y. et al. (2001) Protein kinase C regulates the phosphorylation and cellular localization of
occludin. J. Biol. Chem. 276, 38480-38486
16 Clarke,H. et al. (2000) Protein kinase C activation leads to dephosphorylation of occludin and tight junction
permeability increase in LLC-PK1 epithelial cell sheets. J. Cell Sci. 113 ( Pt 18), 3187-3196
- 32 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
17 Sakakibara,A. et al. (1997) Possible involvement of phosphorylation of occludin in tight junction formation. J.
Cell Biol. 137, 1393-1401
18 Tsukamoto,T. and Nigam,S.K. (1999) Role of tyrosine phosphorylation in the reassembly of occludin and other
tight junction proteins. Am. J. Physiol 276, F737-F750
19 Saitou,M. et al. (1998) Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells
bearing tight junctions. J. Cell Biol. 141, 397-408
20 Saitou,M. et al. (2000) Complex phenotype of mice lacking occludin, a component of tight junction strands.
Mol. Biol. Cell 11, 4131-4142
21 Barrios-Rodiles,M. et al. (2005) High-throughput mapping of a dynamic signaling network in mammalian
cells. Science 307, 1621-1625
22 Furuse,M. et al. (2001) Conversion of zonulae occludentes from tight to leaky strand type by introducing
claudin-2 into Madin-Darby canine kidney I cells. J. Cell Biol. 153, 263-272
23 Inai,T. et al. (1999) Claudin-1 contributes to the epithelial barrier function in MDCK cells. Eur. J. Cell Biol.
78, 849-855
24 Van Itallie,C. et al. (2001) Regulated expression of claudin-4 decreases paracellular conductance through a
selective decrease in sodium permeability. J. Clin. Invest 107, 1319-1327
25 Zeissig,S. et al. (2007) Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight
junctions and barrier dysfunction in active Crohn's disease. Gut 56, 61-72
26 Huber,D. et al. (2000) Occludin modulates transepithelial migration of neutrophils. J. Biol. Chem. 275, 57735778
27 Laukoetter,M.G. et al. (2007) JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp.
Med. 204, 3067-3076
28 Vetrano,S. et al. (2008) Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in
inflammatory bowel disease. Gastroenterology 135, 173-184
29 Fanning,A.S. et al. (1998) The tight junction protein ZO-1 establishes a link between the transmembrane
protein occludin and the actin cytoskeleton. J. Biol. Chem. 273, 29745-29753
30 Itoh,M. et al. (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with
the COOH termini of claudins. J. Cell Biol. 147, 1351-1363
31 Wittchen,E.S. et al. (1999) Protein interactions at the tight junction. Actin has multiple binding partners, and
ZO-1 forms independent complexes with ZO-2 and ZO-3. J. Biol. Chem. 274, 35179-35185
32 Woods,D.F. and Bryant,P.J. (1993) ZO-1, DlgA and PSD-95/SAP90: homologous proteins in tight, septate and
synaptic cell junctions. Mech. Dev. 44, 85-89
33 Haskins,J. et al. (1998) ZO-3, a novel member of the MAGUK protein family found at the tight junction,
interacts with ZO-1 and occludin. J. Cell Biol. 141, 199-208
- 33 -
Chapter 1
34 Meyer,T.N. et al. (2002) Zonula occludens-1 is a scaffolding protein for signaling molecules. Galpha(12)
directly binds to the Src homology 3 domain and regulates paracellular permeability in epithelial cells. J. Biol.
Chem. 277, 24855-24858
35 Balda,M.S. and Matter,K. (2000) The tight junction protein ZO-1 and an interacting transcription factor
regulate ErbB-2 expression. EMBO J. 19, 2024-2033
36 Gottardi,C.J. et al. (1996) The junction-associated protein, zonula occludens-1, localizes to the nucleus before
the maturation and during the remodeling of cell-cell contacts. Proc. Natl. Acad. Sci. U. S. A 93, 10779-10784
37 Poritz,L.S. et al. (2007) Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J.
Surg. Res. 140, 12-19
38 Kucharzik,T. et al. (2001) Neutrophil transmigration in inflammatory bowel disease is associated with
differential expression of epithelial intercellular junction proteins. American Journal of Pathology 159, 2001-2009
39 Anderson,J.M. and Van Itallie,C.M. (1995) Tight junctions and the molecular basis for regulation of
paracellular permeability. Am. J. Physiol 269, G467-G475
40 Hecht,G. et al. (1996) Expression of the catalytic domain of myosin light chain kinase increases paracellular
permeability. Am. J. Physiol 271, C1678-C1684
41 Turner,J.R. et al. (1999) PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC
kinase. Am. J. Physiol 277, C554-C562
42 Hornquist,C.E. et al. (1997) G(alpha)i2-deficient mice with colitis exhibit a local increase in memory CD4+ T
cells and proinflammatory Th1-type cytokines. J. Immunol. 158, 1068-1077
43 Ohman,L. et al. (2000) Immune activation in the intestinal mucosa before the onset of colitis in Galphai2deficient mice. Scand. J. Immunol. 52, 80-90
44 Ohman,L. et al. (2002) Regression of Peyer's patches in G alpha i2 deficient mice prior to colitis is associated
with reduced expression of Bcl-2 and increased apoptosis. Gut 51, 392-397
45 Rudolph,U. et al. (1995) Gi2 alpha protein deficiency: a model of inflammatory bowel disease. J. Clin.
Immunol. 15, 101S-105S
46 Rudolph,U. et al. (1995) Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat.
Genet. 10, 143-150
47 Anderson,J.M. and Van Itallie,C.M. (1995) Tight junctions and the molecular basis for regulation of
paracellular permeability. Am. J. Physiol 269, G467-G475
48 Hecht,G. et al. (1996) Expression of the catalytic domain of myosin light chain kinase increases paracellular
permeability. Am. J. Physiol 271, C1678-C1684
49 Turner,J.R. et al. (1999) PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC
kinase. Am. J. Physiol 277, C554-C562
50 Zhang,Q. et al. (2010) Enteropathogenic Escherichia coli changes distribution of occludin and ZO-1 in tight
junction membrane microdomains in vivo. Microb. Pathog. 48, 28-34
- 34 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
51 Wine,E. et al. (2009) Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional complexes in
polarized epithelia. BMC. Microbiol. 9, 180
52 Kohler,H. et al. (2007) Salmonella enterica serovar Typhimurium regulates intercellular junction proteins and
facilitates transepithelial neutrophil and bacterial passage. Am. J. Physiol Gastrointest. Liver Physiol 293, G178G187
53 Chen,M.L. et al. (2006) Disruption of tight junctions and induction of proinflammatory cytokine responses in
colonic epithelial cells by Campylobacter jejuni. Infect. Immun. 74, 6581-6589
54 Fabia,R. et al. (1993) Impairment of bacterial flora in human ulcerative colitis and experimental colitis in the
rat. Digestion 54, 248-255
55 Favier,C. et al. (1997) Fecal beta-D-galactosidase production and Bifidobacteria are decreased in Crohn's
disease. Dig. Dis. Sci. 42, 817-822
56 Swidsinski,A. et al. (2002) Mucosal flora in inflammatory bowel disease. Gastroenterology 122, 44-54
57 Al Sadi,R. et al. (2008) Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction
permeability. J. Immunol. 180, 5653-5661
58 Fais,S. et al. (1994) Interferon expression in Crohn's disease patients: increased interferon-gamma and -alpha
mRNA in the intestinal lamina propria mononuclear cells. J. Interferon Res. 14, 235-238
59 MacDonald,T.T. et al. (1990) Tumour necrosis factor-alpha and interferon-gamma production measured at the
single cell level in normal and inflamed human intestine. Clin. Exp. Immunol. 81, 301-305
60 Bruewer,M. et al. (2003) Proinflammatory cytokines disrupt epithelial barrier function by apoptosisindependent mechanisms. J. Immunol. 171, 6164-6172
61 Madara,J.L. and Stafford,J. (1989) Interferon-gamma directly affects barrier function of cultured intestinal
epithelial monolayers. J. Clin. Invest 83, 724-727
62 Mullin,J.M. et al. (1992) Modulation of tumor necrosis factor-induced increase in renal (LLC-PK1)
transepithelial permeability. Am. J. Physiol 263, F915-F924
63 Taylor,C.T. et al. (1998) Autocrine regulation of epithelial permeability by hypoxia: role for polarized release
of tumor necrosis factor alpha. Gastroenterology 114, 657-668
64 Zolotarevsky,Y. et al. (2002) A membrane-permeant peptide that inhibits MLC kinase restores barrier function
in in vitro models of intestinal disease. Gastroenterology 123, 163-172
65 Al Sadi,R.M. and Ma,T.Y. (2007) IL-1beta causes an increase in intestinal epithelial tight junction
permeability. J. Immunol. 178, 4641-4649
66 Brun,P. et al. (2007) Increased intestinal permeability in obese mice: new evidence in the pathogenesis of
nonalcoholic steatohepatitis. Am. J. Physiol Gastrointest. Liver Physiol 292, G518-G525
67 Hardin,J. et al. (2000) Effect of proinflammatory interleukins on jejunal nutrient transport. Gut 47, 184-191
68 Matysiak-Budnik,T. et al. (2001) Alterations of epithelial permeability by Helicobacter and IL-1beta in vitro:
protective effect of rebamipide. Dig. Dis. Sci. 46, 1558-1566
- 35 -
Chapter 1
69 Ma,T.Y. et al. (2004) TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires
NF-kappa B activation. Am. J. Physiol Gastrointest. Liver Physiol 286, G367-G376
70 Rescigno,M. et al. (2001) Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers
to sample bacteria. Nature Immunology 2, 361-367
71 Niess,J.H. et al. (2005) CX(3)CR1-mediated dendritic cell access to the intestinal lumen and bacterial
clearance. Science 307, 254-258
72 Rimoldi,M. et al. (2005) Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells
and dendritic cells. Nat. Immunol. 6, 507-514
73 van Niel,G. et al. (2001) Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 121, 337349
74 MacDonald,K.P.A. et al. (2002) Characterization of human blood dendritic cell subsets. Blood 100, 4512-4520
75 Liu,Y.J. et al. (2001) Dendritic cell lineage, plasticity and cross-regulation. Nature Immunology 2, 585-589
76 Grouard,G. et al. (1997) The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3
and CD40-ligand. Journal of Experimental Medicine 185, 1101-1111
77 Rissoan,M.C. et al. (1999) Reciprocal control of T helper cell and dendritic cell differentiation. Science 283,
1183-1186
78 te Velde,A.A. et al. (2003) Increased expression of DC-SIGN(+)IL-12(+)IL-18(+) and CD83(+)IL-12(-)IL-18() dendritic cell populations in the colonic mucosa of patients with Crohn's disease. European Journal of
Immunology 33, 143-151
79 Verstege,M.I. et al. (2008) Dendritic cell populations in colon and mesenteric lymph nodes of patients with
Crohn's disease. J. Histochem. Cytochem. 56, 233-241
80 Janeway,C.A. and Medzhitov,R. (2002) Innate immune recognition. Annual Review of Immunology 20, 197216
81 Hoshino,K. et al. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to
lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162, 3749-3752
82 Poltorak,A. et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4
gene. Science 282, 2085-2088
83 Qureshi,S.T. et al. (1999) Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med.
189, 615-625
84 Wright,S.D. et al. (1989) Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for
recognition by a novel receptor on macrophages. J. Exp. Med. 170, 1231-1241
85 Wright,S.D. et al. (1990) CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding
protein. Science 249, 1431-1433
86 Haziot,A. et al. (1996) Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in
CD14-deficient mice. Immunity. 4, 407-414
- 36 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
87 Braat,H. et al. (2004) Consequence of functional Nod2- and Tlr4-mutations on gene transcription in Crohn's
disease. Gastroenterology 126, A151
88 Takeuchi,O. et al. (2000) Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide
macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent
signaling pathway. J. Immunol. 164, 554-557
89 Takeuchi,O. et al. (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial
lipoproteins. J. Immunol. 169, 10-14
90 Hayashi,F. et al. (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5.
Nature 410, 1099-1103
91 Gewirtz,A.T. et al. (2001) Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce
epithelial proinflammatory gene expression. J. Immunol. 167, 1882-1885
92 Vijay-Kumar,M. et al. (2007) Deletion of TLR5 results in spontaneous colitis in mice. Journal of Clinical
Investigation 117, 3909-3921
93 Gewirtz,A.T. et al. (2006) Dominant-negative TLR5 polymorphism reduces adaptive immune response to
flagellin and negatively associates with Crohn's disease. Am. J. Physiol Gastrointest. Liver Physiol 290, G1157G1163
94 Lodes,M.J. et al. (2004) Bacterial flagellin is a dominant antigen in Crohn disease. J. Clin. Invest 113, 12961306
95 Vijay-Kumar,M. et al. (2007) Activation of Toll-like receptor 3 protects against DSS-induced acute colitis.
Inflammatory Bowel Diseases 13, 856-864
96 Heil,F. et al. (2004) Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science
303, 1526-1529
97 Christensen,S.R. et al. (2006) Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing
inflammatory and regulatory roles in a murine model of lupus. Immunity. 25, 417-428
98 Ghosh,T.K. et al. (2006) Toll-like receptor (TLR) 2-9 agonists-induced cytokines and chemokines: I.
Comparison with T cell receptor-induced responses. Cell Immunol. 243, 48-57
99 Jurk,M. et al. (2006) Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich
phosphorothiate oligodeoxynucleotides. Eur. J. Immunol. 36, 1815-1826
100 Krieg,A.M. (2007) TLR9 and DNA 'feel' RAGE. Nat. Immunol. 8, 475-477
101 Tian,J. et al. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is
mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487-496
102 Lee,J. et al. (2006) Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal
epithelial cells. Nat. Cell Biol. 8, 1327-1336
103 Schlueter,C. et al. (2003) Tissue-specific expression patterns of the RAGE receptor and its soluble forms--a
result of regulated alternative splicing? Biochim. Biophys. Acta 1630, 1-6
- 37 -
Chapter 1
104 Yonekura,H. et al. (2003) Novel splice variants of the receptor for advanced glycation end-products expressed
in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury.
Biochem. J. 370, 1097-1109
105 Medzhitov,R. et al. (1998) MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways.
Molecular Cell 2, 253-258
106 Wesche,H. et al. (1997) MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7,
837-847
107 Araki,A. et al. (2005) MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate
colitis. J. Gastroenterol. 40, 16-23
108 Kawai,T. et al. (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115-122
109 Cao,Z.D. et al. (1996) IRAK: A kinase associated with the interleukin-1 receptor. Science 271, 1128-1131
110 Swantek,J.L. et al. (2000) IL-1 receptor-associated kinase modulates host responsiveness to endotoxin.
Journal of Immunology 164, 4301-4306
111 Lomaga,M.A. et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and
LPS signaling. Genes & Development 13, 1015-1024
112 Nenci,A. et al. (2007) Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446,
557-561
113 Chamaillard,M. et al. (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan
containing diaminopimelic acid. Nat. Immunol. 4, 702-707
114 Inohara,N. et al. (2003) Host recognition of bacterial muramyl dipeptide mediated through NOD2. Journal of
Biological Chemistry 278, 5509-5512
115 Kobayashi,K.S. et al. (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal
tract. Science 307, 731-734
116 Maeda,S. et al. (2005) Nod2 mutation in Crohn's disease potentiates NF-kappa B activity and IL-10
processing. Science 307, 734-738
117 Watanabe,T. et al. (2004) NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1
responses. Nature Immunology 5, 800-808
118 Hugot,J.P. et al. (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's
disease. Nature 411, 599-603
119 Ogura,Y. et al. (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease.
Nature 411, 603-606
120 Miceli-Richard,C. et al. (2001) CARD15 mutations in Blau syndrome. Nat. Genet. 29, 19-20
121 Martinon,F. et al. (2004) Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin
inflammasome. Curr. Biol. 14, 1929-1934
- 38 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
122 Hoffman,H.M. et al. (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial
cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301-305
123 Villani,A.C. et al. (2009) Common variants in the NLRP3 region contribute to Crohn's disease susceptibility.
Nat. Genet. 41, 71-76
124 Dinarello,C.A. (1998) Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann.
N. Y. Acad. Sci. 856, 1-11
125 Agostini,L. et al. (2004) NALP3 forms an IL-1beta-processing inflammasome with increased activity in
Muckle-Wells autoinflammatory disorder. Immunity. 20, 319-325
126 Reimund,J.M. et al. (1996) Increased production of tumour necrosis factor-alpha interleukin-1 beta, and
interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn's disease. Gut 39, 684-689
127 Ten Hove,T. et al. (2001) Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing
local TNF-alpha production in mice. Gastroenterology 121, 1372-1379
128 Drickamer,K. (1999) C-type lectin-like domains. Current Opinion in Structural Biology 9, 585-590
129 Figdor,C.G. et al. (2002) C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol.
2, 77-84
130 Robinson,M.J. et al. (2006) Myeloid C-type lectins in innate immunity. Nat. Immunol. 7, 1258-1265
131 Cambi,A. and Figdor,C.G. (2003) Dual function of C-type lectin-like receptors in the immune system.
Current Opinion in Cell Biology 15, 539-546
132 Garcia-Vallejo,J.J. and van,K.Y. (2009) Endogenous ligands for C-type lectin receptors: the true regulators of
immune homeostasis. Immunol. Rev. 230, 22-37
133 Geijtenbeek,T.B. and Gringhuis,S.I. (2009) Signalling through C-type lectin receptors: shaping immune
responses. Nat. Rev. Immunol. 9, 465-479
134 Cambi,A. et al. (2003) The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida
albicans on dendritic cells. European Journal of Immunology 33, 532-538
135 Geijtenbeek,T.B.H. and van Kooyk,Y. (2003) DC-SIGN: A novel HIV receptor on DCs that mediates HIV-1
transmission. Dendritic Cells and Virus Infection 276, 31-54
136 Geijtenbeek,T.B.H. et al. (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. Journal of
Experimental Medicine 197, 7-17
137 Lozach,P.Y. et al. (2003) DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus
glycoprotein E2. Journal of Biological Chemistry 278, 20358-20366
138 Tassaneetrithep,B. et al. (2003) DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells.
Journal of Experimental Medicine 197, 823-829
139 Geijtenbeek,T.B. et al. (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med.
197, 7-17
- 39 -
Chapter 1
140 Caparros,E. et al. (2006) DC-SIGN ligation on dendritic cells results in ERK and PI3K activation and
modulates cytokine production. Blood 107, 3950-3958
141 Nagaoka,K. et al. (2005) Association of SIGNR1 with TLR4-MD-2 enhances signal transduction by
recognition of LPS in gram-negative bacteria. International Immunology 17, 827-836
142 Brown,G.D. et al. (2003) Dectin-1 mediates the biological effects of beta-glucans. Journal of Experimental
Medicine 197, 1119-1124
143 Gantner,B.N. et al. (2003) Collaborative induction of inflammatory responses by dectin-1 and toll-like
receptor 2. Journal of Experimental Medicine 197, 1107-1117
144 Banchereau,J. et al. (2000) Immunobiology of dendritic cells. Annual Review of Immunology 18, 767-+
145 Braegger,C.P. et al. (1992) Tumour necrosis factor alpha in stool as a marker of intestinal inflammation.
Lancet 339, 89-91
146 Nicholls,S. et al. (1993) Cytokines in stools of children with inflammatory bowel disease or infective
diarrhoea. J. Clin. Pathol. 46, 757-760
147 Kontoyiannis,D. et al. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich
elements: implications for joint and gut-associated immunopathologies. Immunity. 10, 387-398
148 Wallach,D. et al. (1997) Cell death induction by receptors of the TNF family: towards a molecular
understanding. FEBS Lett. 410, 96-106
149 Grell,M. et al. (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the
80 kDa tumor necrosis factor receptor. Cell 83, 793-802
150 Starr,T.K. et al. (2003) Positive and negative selection of T cells. Annual Review of Immunology 21, 139-176
151 Bach,J.F. (2003) Regulatory T cells under scrutiny. Nature Reviews Immunology 3, 189-198
152 Shevach,E.M. et al. (2001) Control of T-cell activation by CD4(+) CD25(+) suppressor T cells.
Immunological Reviews 182, 58-67
153 Bluestone,J.A. and Abbas,A.K. (2003) Natural versus adaptive regulatory T cells. Nature Reviews
Immunology 3, 253-257
154 Cavani,A. et al. (2000) Human CD4(+) T lymphocytes with remarkable regulatory functions on dendritic cells
and nickel-specific Th1 immune responses. Journal of Investigative Dermatology 114, 295-302
155 Khoo,U.Y. et al. (1997) CD4(+) T cell down-regulation in human intestinal mucosa - Evidence for intestinal
tolerance to luminal bacterial antigens. Journal of Immunology 158, 3626-3634
156 Fukaura,H. et al. (1996) Induction of circulating myelin basic protein and proteolipid protein-specific
transforming growth factor-beta 1-secreting Th3 T cells by oral administration of myelin in multiple sclerosis
patients. Journal of Clinical Investigation 98, 70-77
157 Inobe,J. et al. (1998) IL-4 is a differentiation factor for transforming growth factor-beta secreting Th3 cells
and oral administration of IL-4 enhances oral tolerance in experimental allergic encephalomyelitis. European
Journal of Immunology 28, 2780-2790
- 40 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
158 Levings,M.K. et al. (2001) IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells.
Journal of Immunology 166, 5530-5539
159 Jonuleit,H. et al. (2000) Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with
regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. Journal of
Experimental Medicine 192, 1213-1222
160 Dhodapkar,M.V. et al. (2001) Antigen-specific inhibition of effector T cell function in humans after injection
of immature dendritic cells. Journal of Experimental Medicine 193, 233-238
161 Smits,H.H. et al. (2005) Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by
modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing
nonintegrin. Journal of Allergy and Clinical Immunology 115, 1260-1267
162 Brady,M.T. et al. (2003) Hepatitis C virus non-structural protein 4 suppresses Th1 responses by stimulating
IL-10 production from monocytes. European Journal of Immunology 33, 3448-3457
163 Lavelle,E.C. et al. (2003) Cholera toxin promotes the induction of regulatory T cells specific for bystander
antigens by modulating dendritic cell activation. Journal of Immunology 171, 2384-2392
164 McGuirk,P. et al. (2002) Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial
molecule that stimulates interleukin 10 production by dendritic cells: A novel strategy for evasion of protective T
helper type 1 responses by Bordetella pertussis. Journal of Experimental Medicine 195, 221-231
165 Ross,P.J. et al. (2004) Adenylate cyclase toxin from Bordetella pertussis synergizes with lipopolysaccharide
to promote innate interleukin-10 production and enhances the induction of Th2 and regulatory T cells. Infection
and Immunity 72, 1568-1579
166 Braat,H. et al. (2007) Prevention of experimental colitis by parenteral administration of a pathogen-derived
immunomodulatory molecule. Gut 56, 351-357
167 Adams,V.C. et al. (2004) Mycobacterium vaccae induces a population of pulmonary CD11c(+) cells with
regulatory potential in allergic mice. European Journal of Immunology 34, 631-638
168 Zuany-Amorim,C. et al. (2002) Suppression of airway eosinophilia by killed Mycobacterium vaccae-induced
allergen-specific regulatory T-cells. Nature Medicine 8, 625-629
169 Hart,A.L. et al. (2004) Modulation of human dendritic cell phenotype and function by probiotic bacteria. Gut
53, 1602-1609
170 Summers,R.W. et al. (2005) Trichuris suis therapy in Crohn's disease. Gut 54, 87-90
171 Braat,H. et al. (2006) A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn's disease.
Clin. Gastroenterol. Hepatol. 4, 754-759
172 Aggarwal,S. et al. (2003) Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the
production of interleukin-17. J. Biol. Chem. 278, 1910-1914
173 Kobayashi,T. et al. (2008) IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's
disease. Gut 57, 1682-1689
- 41 -
Chapter 1
174 Langrish,C.L. et al. (2005) IL-23 drives a pathogenic T cell population that induces autoimmune
inflammation. J. Exp. Med. 201, 233-240
175 Duerr,R.H. et al. (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease
gene. Science 314, 1461-1463
176 Elson,C.O. et al. (2007) Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in
mice. Gastroenterology 132, 2359-2370
177 Moschen,A.R. et al. (2008) Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology
213, 779-787
178 Ouyang,W. et al. (2008) The biological functions of T helper 17 cell effector cytokines in inflammation.
Immunity. 28, 454-467
179 Fina,D. et al. (2008) Regulation of gut inflammation and th17 cell response by interleukin-21.
Gastroenterology 134, 1038-1048
180 Fantini,M.C. et al. (2007) IL-21 regulates experimental colitis by modulating the balance between Treg and
Th17 cells. Eur. J. Immunol. 37, 3155-3163
181 Mucida,D. et al. (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid.
Science 317, 256-260
182 Xiao,S. et al. (2008) Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells
by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol.
181, 2277-2284
183 Cosmi,L. et al. (2008) Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor.
J. Exp. Med. 205, 1903-1916
184 Kleinschek,M.A. et al. (2009) Circulating and gut-resident human Th17 cells express CD161 and promote
intestinal inflammation. J. Exp. Med. 206, 525-534
185 Bassotti,G. et al. (2007) Enteric glial cells: new players in gastrointestinal motility? Lab Invest 87, 628-632
186 Savidge,T.C. et al. (2007) Starring roles for astroglia in barrier pathologies of gut and brain. Lab Invest 87,
731-736
187 de Jonge,W.J. et al. (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the
Jak2-STAT3 signaling pathway. Nat. Immunol. 6, 844-851
188 Ghia,J.E. et al. (2006) The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel
disease in a murine model. Gastroenterology 131, 1122-1130
189 Luyer,M.D. et al. (2005) Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the
vagus nerve. J. Exp. Med. 202, 1023-1029
190 Mioni,C. et al. (2005) Activation of an efferent cholinergic pathway produces strong protection against
myocardial ischemia/reperfusion injury in rats. Crit Care Med. 33, 2621-2628
- 42 -
Intestinal interactions between the epithelial barrier, immune system and nervous system
191 The,F.O. et al. (2007) Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative
ileus in mice. Gastroenterology 133, 1219-1228
192 van Westerloo,D.J. et al. (2005) The cholinergic anti-inflammatory pathway regulates the host response
during septic peritonitis. J. Infect. Dis. 191, 2138-2148
193 van Westerloo,D.J. et al. (2006) The vagus nerve and nicotinic receptors modulate experimental pancreatitis
severity in mice. Gastroenterology 130, 1822-1830
194 Ghia,J.E. et al. (2007) The protective effect of the vagus nerve in a murine model of chronic relapsing colitis.
Am. J. Physiol Gastrointest. Liver Physiol 293, G711-G718
195 Borovikova,L.V. et al. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to
endotoxin. Nature 405, 458-462
196 Matsunaga,K. et al. (2001) Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial
activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J.
Immunol. 167, 6518-6524
197 Wang,H. et al. (2004) Cholinergic agonists inhibit HMGB1 release and improve survival in experimental
sepsis. Nat. Med. 10, 1216-1221
198 Pullan,R.D. et al. (1994) Transdermal nicotine for active ulcerative colitis. N. Engl. J. Med. 330, 811-815
199 Sandborn,W.J. et al. (1997) Transdermal nicotine for mildly to moderately active ulcerative colitis. A
randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 126, 364-371
200 Arranz,A. et al. (2008) Vasoactive intestinal peptide as a healing mediator in Crohn's disease.
Neuroimmunomodulation. 15, 46-53
201 Gareau,M.G. et al. (2007) Neonatal maternal separation of rat pups results in abnormal cholinergic regulation
of epithelial permeability. Am. J. Physiol Gastrointest. Liver Physiol 293, G198-G203
202 Greenwood,B. and Mantle,M. (1992) Mucin and protein release in the rabbit jejunum: effects of bethanechol
and vagal nerve stimulation. Gastroenterology 103, 496-505
203 Neunlist,M. et al. (2003) Human ENS regulates the intestinal epithelial barrier permeability and a tight
junction-associated protein ZO-1 via VIPergic pathways. Am. J. Physiol Gastrointest. Liver Physiol 285, G1028G1036
204 Toumi,F. et al. (2003) Human submucosal neurones regulate intestinal epithelial cell proliferation: evidence
from a novel co-culture model. Neurogastroenterol. Motil. 15, 239-242
205 Cameron,H.L. and Perdue,M.H. (2007) Muscarinic acetylcholine receptor activation increases transcellular
transport of macromolecules across mouse and human intestinal epithelium in vitro. Neurogastroenterol. Motil.
19, 47-56
206 Savidge,T.C. et al. (2007) Enteric glia regulate intestinal barrier function and inflammation via release of Snitrosoglutathione. Gastroenterology 132, 1344-1358
207 Neunlist,M. et al. (2007) Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1dependent pathway. Am. J. Physiol Gastrointest. Liver Physiol 292, G231-G241
- 43 -
Chapter 1
- 44 -
Part 1
Apoptosis and soluble
tumour necrosis factor
inhibitors in inflammatory
bowel diseases
Chapter
2.
Apoptosis as a therapeutic paradigm in
inflammatory bowel diseases
M.I. Verstege1, A.A. te Velde1, D.W. Hommes2
1 Centre for Experimental and Molecular Medicine, Academic Medical Centre,
Amsterdam, the Netherlands
2 Department of Gastroenterology and Hepatology, Leiden University Medical
Centre, Leiden, the Netherlands
Chapter 2
Abstract
Evidence is increasing that a defect in apoptosis is involved in the pathogenesis of
inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis
(UC). CD seems to be the cause of an intrinsic defect in the apoptotic pathway of
(autoreactive) T cells, resulting in excessive T cell responses. In UC, an increased rate of
apoptosis of epithelial cells is observed. In this review we will describe apoptotic
mechanisms and their association to IBD. In addition, we will review how specific
therapeutic approaches interact at different levels with the apoptotic pathway.
- 48 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
Introduction
Inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis
(UC) are chronic inflammatory disorders of the intestinal tract with an unknown aetiology.
Although the pathogenesis of IBD is not well understood, evidence is increasing that
excessive mucosal T cell-dependent responses in IBD are due to a defect in apoptosis.
Muramyl dipeptide stimulated monocyte-derived dendritic cells from CD carriers of
double-dose NOD2 mutations demonstrated a change in gene expression profiles resulting
in a negative regulation of apoptosis 1.
Apoptosis or programmed cell death is a normal physiological process, which
plays an important role in the development and morphogenesis, cellular homeostasis and
the deletion of damaged and autoreactive cells, e.g. the formation of the digits 2, the
development of the brain 3 and reproductive organs 4, the negative selection of T cells in the
thymus 5 and the deletion of infected and cancer cells 6. In the gut, apoptosis is important in
the homeostasis of the epithelium by regulating cell numbers of epithelial cells and in the
elimination of lamina propria T lymphocytes (LPLs) to prevent an excessive immune
response because of the constitutive examination of luminal antigens 7,8.
In contrast to necrosis, apoptosis will not lead to an inflammatory reaction,
because of a tightly and controlled process in which the cell shrinks, the chromatin
condensates and marginates at the nuclear membrane and ‘budding’ of the plasma
membrane will result in apoptotic bodies that contain cytoplasm and organelles, which can
be phagocytosed by immune cells 9. Necrotic cells, however, are swollen and leaky and
finally, because of cellular and nuclear lysis, the complete contents of the cells will be
released in the surrounding tissues, resulting in inflammatory reactions 10.
- 49 -
Chapter 2
The cell cycle
The cell cycle is regulated by a large family of enzymes mentioned as cyclin-dependent
kinases (CDKs), which play a key role in the phosphorylation and consequently activation
of downstream molecules that are involved in DNA replication and mitosis (see figure 1).
CDKs are activated by association with cyclins and are inhibited by other kinases and
phosphatases such as p15, p16, p21 and p27. In a controlled cell cycle, the interaction
between CDKs, cylins and CDK inhibitors is tightly regulated to induce cell growth when
necessary, but to prevent excessive cell growth that could result in tumourgenesis. The cell
cycle suppressor proteins p15 and p27 could be activated by transforming growth factor
(TGF)-, which plays an important role in the inhibition of epithelial cell proliferation 11,12.
It has been shown that TGF- expression is increased in murine LPLs and colonic epithelial
cells and that epithelial apoptosis is increased in mice or rats that developed colitis by oral
administration of dextran sodium sulphate (DSS) 13,14. These results indicate that epithelial
cells and lymphocytes in UC becomes more sensitive to apoptosis by a constitutively
inhibition of cell growth. In human UC patients, however, the results are not so clear.
Different studies have demonstrated that apoptosis of LPLs in both in UC as well as CD
patients is decreased
7,15,16
. On the contrary, several other studies have shown that LPLs of
UC patients have a delayed cell cycle progression leading to increased apoptosis of these
lymphocytes
17-19
. Moreover, apoptosis of epithelial cells, mainly located at the apical
surface of the mucosa is increased in UC patients compared to normal controls 17,20. It could
be hypothesized that increased apoptosis of epithelial cells increases mucosal exposure to
intestinal bacteria inducing an inflammatory response in UC patients. This hypothesis is
still speculative, but the treatment of IL-10 deficient mice with the non-steroidal antiinflammatory drug piroxicam has been shown to enhance apoptosis of epithelial cells in
combination with a decreased barrier function and acceleration of the development of
colitis 21.
In response to cellular stress, e.g. DNA damage and hypoxia, the tumour
suppressor gene p53 becomes activated by phosphorylation resulting in the activation of the
CDK inhibiter p21, which will finally lead to apoptosis 22. Stabilisation and activation of
- 50 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
p53 is initiated by ARF, which blocks the Mdm2-mediated suppression of p53. Mdm-2
mediates ubiquitination of p53, which will be degenerated by the proteasome 23. Binding of
ARF inhibits the ubiquitin-ligase activity of Mdm-2, thereby increasing active p53 levels 24.
ARF expression is mediated by the transcription factor E2F, which is activated by several
oncogenes, e.g. ras and myc, and inhibited by the tumour suppressor gene product
retinoblastoma (Rb)
25
. It has been demonstrated that LPLs of CD patients have an
increased expression of phosphorylated Rb and a decreased expression of phosphorylated
p53, both resulting in resistance to apoptosis and dynamic cellular expansion
19
. On the
26
contrary, p53 is overexpressed in areas of acute inflammation in UC patients .
- 51 -
Chapter 2
Figure 1. The cell cycle. The cell cycle is tightly regulated by CDKs, which are activated by association with
cyclins and are inhibited by p15, p16, p21 and p27. TGF- is a suppressor of p15 and p27 and consequently
inhibits epithelial cell proliferation. In response to cellular stress, p53 becomes activated resulting in the activation
of the CDK inhibitor p21, the activation of the pro-apoptotic proteins Bax and Apaf-1 and the inhibition of the
anti-apoptotic proteins Bcl-2 and Bcl-XL, finally leading to apoptosis. Mdm-2 mediates ubiquitination of p53 so
that it will be degenerated by the proteasome. ARF is able to stabilise and activate p53 by blocking the Mdm2mediated suppression of p53. The expression of ARF is mediated by E2F which is activated by several oncogenes,
including ras and myc and is inhibited by the tumour suppressor product Rb. Oncogenes also activate Akt leading
to Mdm2 activation
- 52 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
Death receptor pathway
Both internal as well as external stimuli are able to induce apoptosis, such as the activation
of different surface molecules, DNA damage caused by defective DNA repair mechanisms
and irradiation, an uncontrolled cell cycle, cytotoxic drugs or a lack of survival signals (see
figure 2). Three major pathways in the initiation of apoptosis have been described: 1) the
death receptor pathway, 2) the mitochondrial pathway and 3) the endoplasmatic reticulum
stress pathway. The death receptor pathway is an extrinsic apoptosis pathway characterised
by the interaction between death receptors and their ligands, the recruitment of adapter
proteins and the activation of several caspases. This classical pathway which is mainly
caspase-dependent is restricted to the so-called type I cells 27. Death receptors belong to a
family of tumour necrosis factor receptors (TNFRs) and nerve growth factor receptors
(NGFRs) of which the Fas- (CD95) and TNFR-mediated apoptosis are the best studied. Fas
is a type I transmembrane receptor that is widely expressed and constitutively expressed by
T lymphocytes, whereas FasL is a type II transmembrane protein that is induced on
activated T lymphocytes. Upon ligation of FasL, Fas forms trimers and through its death
domain (DD) recruitment of the adapter molecule Fas-associated death domain (FADD) is
achieved. FADD contains death effecter domains (DEDs) that recruit and binds to procaspase-8 (flice) to form the death-inducing signalling complex (DISC) 28. Pro-caspase-8 is
activated by autoproteolytic cleavage of caspase-8, which consists of two heterodimers of
two small and two large subunits
29
. Active caspase-8 cleaves downstream effector pro-
caspases in active caspases that cleave several transcription factors, structural proteins and
enzymes involved in DNA repair and cleavage and cell cycle progression, finally resulting
in cell death. Neutrophils isolated from both CD as well as UC patients exhibit a decreased
expression of pro-caspase-3, one of the substrates of caspase 8, leading to delayed apoptosis
30
. The Fas-mediated apoptosis can be inhibited by Fas-associate-death domain-like
interleukin-1-converting enzyme (Flice)-inhibitory protein (Flip), which is an intracellular
protein that prevents the cleavage of pro-caspase-8 31,32. The expression of Flip is enhanced
by LPLs from CD compared to normal controls, resulting in resistance to apoptosis 19,33. In
addition, in CD patients the expression of Fas is reduced in T cells and macrophages
located in the LP in situ, whereas rates of FasL-expressing cells in the LP are increased,
- 53 -
Chapter 2
indicating that LPLs of CD patients are less sensitive to extrinsic apoptotic signals
17
.
Isolated LPLs from CD patients that were cultured with a CD2 activation pathway stimulus
express similar levels of Fas compared to controls; however they are less sensitive to Fasmediated apoptosis induced by Fas antigen cross linking
34
. Also in UC there are
indications that the Fas-ligand mediated apoptosis is disturbed in LPLs, although some of
the results are contradictive. Suzuki et al. have shown that the expression of FasL is
increased by memory CD4+ LPLs, but that the expression of Fas is not different compared
to normal controls
15
. Since the apoptotic cell ratio induced by anti-Fas antibody did not
change, it seems that memory CD4+ LPLs are less sensitive to Fas-ligand mediated
apoptosis in UC patients. However, Strater et al. have demonstrated that in UC patients, the
expression of both FasL as well as Fas is increased by LPLs, indicating that LPLs of UC
patients are more sensitive to apoptosis
18
. Moreover, they have shown that also colonic
epithelial cells have an increased sensitivity to apoptosis
. Besides, Yukawa et al. have
18
demonstrated that in active UC, an increased number of FasL-expressing cells was present
in the mucosa and that the number of apoptotic cells was increased compared to UC
patients in remission, non-IBD colitis and controls 20. In conclusion, LPLs of CD patients
are less sensitive to Fas-mediated apoptosis, whereas Fas-mediated apoptosis sensitivity
seems to be increased in LPLs and epithelial cells of UC patients.
Besides Fas, apoptosis can also be induced through the TNFRs upon binding to its
ligand TNF-. TNFRs are type I transmembrane proteins of which only TNFR-I contains a
DD. Since TNFR-II lacks a DD, this molecule is mainly involved the survival of the cell,
whereas TNFR-I activates both cell survival pathways as well as death signalling. Upon
ligation with TNF-, TNFR-I forms trimers via its DD, followed by the recruitment of the
adapter molecule TNFR-associated DD (TRADD) and FADD. FADD recruits pro-caspase8 and activation of this molecule by dimerisation results in the cleavage of downstream
effector caspases and finally in apoptosis, a pathway similar as described for Fas-mediated
apoptosis. Conversely, FADD is able to activate cell survival pathways as well by the
recruitment of TRAF-2 and RIP, molecules that induce pathways leading to mitogenactivated protein kinase (MAPK) and NF-B, respectively. NF-B is a transcription factor
that induces the expression of pro-inflammatory genes, including TNF-, IL-1, IL-6 and
- 54 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
IL-12, and anti-apoptotic genes such as Flip, Traf-1, Bcl-2 and Bcl-xl (see below).
Bregenholt et al. demonstrated that apoptosis of LPLs is mainly induced via the Fasmediated pathway and not via the TNF- receptor in CD4+ -transferred SCID mice with
colitis 35.
Since TNFR-II does not harbour a DD, they are not able to induce apoptosis via
FADD, but there are some data that also TNFR-II can induce TNF--mediated apoptosis 3639
. Nevertheless, over-expression of TNFR-II promotes colitis in SCID mice transferred
with CD4+CD62L+ T cells characterised by aggravated Th1 response and apoptotic
resistance, indicating that TNFR-II plays a prominent role in cell survival 40. In addition, it
has been shown that TNFR-II expression by LPLs and peripheral blood T lymphocytes is
increased in CD patients, whereas TNFR-I levels were similar to normal controls 40.
- 55 -
Chapter 2
Figure 2. Apoptotic signalling. The death receptor pathway is characterised by the activation of Fas by FasL
resulting in the recruitment of FADD and pro-caspase-8 to form the death-inducing signalling complex (DISC).
Pro-caspase is cleaved in caspase-8, which cleaves other downstream pro-caspases into their active form,
including pro-caspase-3, leading to apoptosis. Apoptosis could be prevented by Flip, since it inhibits the cleavage
of pro-caspase-8. This pathway can be amplified through the mitochondrial pathway by the cleavage of Bid by
caspase-8. Also stimuli such as DNA damage are able to activate the mitochondrial pathway. DNA damage leads
to the activation of p53 and consequently of the activation of pro-apoptotic proteins Bak and Bax. Overexpression
of Bcl-2 and Bcl-XL prevents activation of Bak and Bax, resulting in cell survival. Bid, Bak and Bax contribute to
permeabilisation of the outer membrane or by interaction with voltage dependent anion channels in the outer
membrane mitochondrion, leading to the release of cytochrome c in the cytoplasm. Cytochrome c forms together
with Apaf-1 and pro-caspase-9 the apoptosome resulting in activated caspase-9, which cleaves pro-caspase-3 into
its active form, leading to downstream activation of other caspases and finally in apoptosis. The apoptosome and
caspase-3 could be inhibited by IAPs, so that apoptosis is prevented.
- 56 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
Mitochondrial pathway
Type II cells are not able to induce apoptosis though the classical pathway since procaspase-8 concentrations are too low to induce a strong enough caspase signalling cascade.
Therefore the signal has to be amplified through the mitochondrial-mediated pathway,
which is activated by the cleavage of Bid by caspase-8. Truncated Bid is capable to
translocate from the cytoplasm into the mitochondria where it induces conformational
changes in the pro-apoptotic protein Bax, resulting in the release of cytochrome c in the
cytoplasm. Cytoplasmic cytochrome c interacts with the adaptor molecule proteaseactivating factor (Apaf)-1 in a dATP-dependent manner to assembly the apoptosome that
initiates the activation of pro-caspase-9. Activated caspase-9 cleaves other downstream
caspases, including caspase-3, -7 and -6, consequently inducing apoptosis.
In addition to activation via the classical pathway, other stimuli such as DNA
damage caused by e.g. irradiation, chemotherapeutic drugs, stress molecules and a lack of
growth factors also can promote mitochondrial-mediated apoptosis. A central role in the
mitochondrial-mediated apoptosis plays the Bcl-2 family of proteins, which harbours both
pro- as well as anti-apoptotic members. Dependent on the presence of the highly conserved
Bcl-2 homology domains (BH1 to BH4), the Bcl-2 family of proteins can be defined into
three groups. The first group is characterised by the presence of all the four domains and by
their anti-apoptotic effects (e.g. Bcl-2 and Bcl-XL). The second and third group, however,
both consist of proteins with pro-apoptotic effects and are distinguished by the presence of
the domains BH1 to BH3 (e.g. Bax, Bak) or only the BH3 domain, respectively (e.g. Bid).
Activation of p53 e.g. due to DNA damage, Bax and Bak are activated, which are thought
to contribute to the permeabilisation of the outer membrane of the mitochondrion
41
or by
the interaction with voltage dependent anion channels in the outer membrane 42, leading to
the release of cytochrome c in the cytoplasm and consequently to apoptosis of the cell.
Overexpression of Bcl-2 and Bcl-XL prevents oligomerisation and activation of Bak and
Bax, which will result in cell survival 43. In healthy colonic tissue of mice it has been shown
that the expression of Bcl-2 is prominent in the base of the crypts, whereas surface
epithelial cells express increased levels of Bax, consistent with cell growth in the bottom of
- 57 -
Chapter 2
the crypts and cell death at the surface 44. However, in both mice and humans, it has been
demonstrated that different components of the mitochondrial pathway are affected in
colitis. Gi2-deficient mice, which spontaneously develop an UC-like disease, exhibit an
increased apoptosis of lymphocytes associated with decreased levels of Bcl-2 leading to
regression of Peyer’s patches, both in number as well as in size 45. Besides, mice which are
deficient for poly-(ADP-ribose) polymerase-1 (PARP-1), which is activated in response to
DNA damage, develop less severe TNBS-induced colitis accompanied with an increased
expression of Bcl-2 and reduced apoptosis of epithelial cells compared to wild type mice 46.
TNBS-induced colitis in wild type mice results in a decrease of the anti-apoptotic protein
Bcl-2, whereas the pro-apoptotic protein Bax remains unchanged
46
. This indicates that
disruption of the epithelial barrier due to a genetic defect or a reagent results in apoptosis of
LPLs and epithelial cells. In human CD patients, however, a decreased Bax to Bcl-2 ratio
accompanied by a decreased level of Bax and/or an increased level of Bcl-2, results in
apoptotic resistance of mucosal T lymphocytes 34,47,48.
- 58 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
Apoptosis as a therapeutic paradigm in IBD
Currently, (gluco-) corticosteroids, thiopurines, methotrexate, 5-amino salicylic acid (5ASA) and its derivates and inhibitors of TNF- are commonly used in IBD. Several of
these compounds interfere at different levels with the apoptotic pathway. Sulphasalazine is
a conjugate of 5-ASA and sulphapyridine and has been shown to inhibit the
phosphorylation of NF-B directly
49
. Since NF-B is not only a transcription factor for
several pro-inflammatory genes, but also for anti-apoptotic genes, the inhibition of NF-B
may lead to an increased susceptibility to apoptosis. Doering et al. demonstrated that
sulphasalazine, however not 5-ASA and sulphapyridine alone, induces apoptosis of LPLs in
a Fas-independent way
16
. Sulphasalazine treatment of Jurkat T cells results in a down-
regulation of the anti-apoptotic molecules Bcl-2 and Bcl-XL and subsequent activation of
caspase-3 and -9, indicating that sulphasalazine acts via the mitochondrial pathway to
induce apoptosis
16
. Another common used drug in the treatment of IBD is azathioprine.
Tiede et al. have demonstrated that azathioprine and its metabolites 6-mercaptopurine and
6-thioguanine nucleotides only induce apoptosis of CD4+ lymphocytes when they are costimulated through CD28 50, suggesting that azathioprine induces apoptosis of activated T
lymphocytes. The TNF- neutralising drugs infliximab and etanercept are both efficacious
in the treatment of rheumatoid arthritis, however; only infliximab has been shown to be
effective in the treatment of CD
51,52
. This suggests different mechanisms in which both
TNF- neutralizing drugs act. Van den Brande et al. have demonstrated that both
infliximab as well as etanercept neutralize soluble TNF- effectively
53
. However,
Infliximab but not etanercept has the capacity to induce apoptosis of activated peripheral
blood lymphocytes, LPLs and monocytes
53,54
. Apoptosis induced by infliximab is
associated with an increase in the Bax/Bcl-2 ratio in Jurkat T cells that are CD3/CD28
stimulated, but not in unstimulated cells 54.
Also potential new therapeutic strategies in IBD correlate with apoptosis. IL-6
seems to play an important role in the maintenance of the intestinal inflammation 55. The
expression of IL-6 and its soluble receptor by LPLs is increased in IBD patients 55 and will
- 59 -
Chapter 2
result to an elevated expression of STAT-3 and its nuclear translocation leading to the
transcription of anti-apoptotic genes, including Bcl-2 and Bcl-xl. Elevated IL-6 expression
will finally lead to an increased resistance to apoptosis of LPLs and is therefore an
interesting target in the treatment of IBD. Both antibodies against the IL-6 receptor as well
as the soluble form have been shown to reduce inflammation processes in several
experimental colitis models
50,56,57
. Treatment with antibodies against the IL-6 receptors
suppresses the mRNA expression of adhesion molecules such as ICAM-1 and VCAM-1
and pro-inflammatory cytokines including IFN-, TNF- and IL-1
56
. Moreover, the
expression of active STAT-3 is reduced, whereas the number of apoptotic LPLs is
increased
. In addition, in vitro studies showed that LPLs of CD patients undergo
57
apoptosis after being treated by antibodies against the IL-6 receptor 50. Antibodies against
the Th1-inducing cytokine IL-12 also reduce experimental colitis and induce apoptosis of
LPLs in a Fas-dependent way, because Fas-deficient or Fas-Fc treated mice are resistant to
anti-IL-12 treatment 58.
NF-B activation is observed in IBD, and a potential new strategy would be to
block this transcription factor by NF-B decoy oligonucleotides
59
. These decoy
+
oligonucleotides induces CD4 T lymphocyte apoptosis and reduces the inflammation in
both the Th1-mediated TNBS-induced colitis model as well as the Th2-mediated
oxazolone-induced colitis 59. Also the use of antisense oligonucleotides to inhibit the antiapoptotic protein Flip restores apoptosis in isolated LPLs 33.
A second approach in the treatment of IBD is the use of plant-derived compounds
that harbour anti-inflammatory and anti-oxidant effects. Garlicin is a compound extracted
from garlic corn and has been shown to be effective in TNBS-induced colitis in rats by
reducing the expression of the anti-apoptotic protein Bcl-2 by lymphocytes resulting in
increased apoptosis
60
. Also resveratrol, which is a polyphenolic compound derived from
grapes and wine, is able to reduce TNBS-induced colitis in rats and to enhance apoptosis 61.
The mechanisms by which these compounds are capable in reducing intestinal
inflammation and enhancing apoptosis have still to be elucidated.
- 60 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
Finally, it is also possible to make use of immunosuppressive capacities of several
micro-organisms to escape the immune system of the host. Gi2-deficient mice develop
less severe colitis when treated with acellular Bordetella pertussis vaccine containing
filamentous haemagglutinin due to an increased IL-10 production in the intestinal mucosa
and an increased apoptosis of Th1 cells 62.
- 61 -
Chapter 2
Conclusion
Although CD and UC are both chronic inflammatory diseases of the intestines their
aetiology and pathogenesis seems to be different. In UC, it seems that an increased
apoptosis of epithelial cells results in the destruction of the epithelial barrier, so that
intestinal components, e.g. bacteria and food particles have excess to the LP where they
induce an inflammatory reaction. On the contrary, CD is probably the cause of an intrinsic
defect in the apoptotic pathway of (autoreactive) T cells. Therefore, new therapeutic
approaches have to focus on the different aspects that are involved in the apoptotic
pathways in UC and CD.
- 62 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
References
1 Zelinkova,Z. et al. (2006) Gene expression profile in Crohn's disease related NOD2 variants. Gastroenterology
130, A-363
2 Zuzarte-Luis,V. and Hurle,J.M. (2002) Programmed cell death in the developing limb. Int. J. Dev. Biol. 46, 871876
3 Hutchins,J.B. and Barger,S.W. (1998) Why neurons die: cell death in the nervous system. Anat. Rec. 253, 79-90
4 Meier,P. et al. (2000) Apoptosis in development. Nature 407, 796-801
5 Rathmell,J.C. and Thompson,C.B. (2002) Pathways of apoptosis in lymphocyte development, homeostasis, and
disease. Cell 109 Suppl, S97-107
6 Fadeel,B. et al. (1999) Apoptosis in human disease: a new skin for the old ceremony? Biochem. Biophys. Res.
Commun. 266, 699-717
7 Bu,P. et al. (2001) Apoptosis: one of the mechanisms that maintains unresponsiveness of the intestinal mucosal
immune system. J. Immunol. 166, 6399-6403
8 Hall,P.A. et al. (1994) Regulation of cell number in the mammalian gastrointestinal tract: the importance of
apoptosis. J. Cell Sci. 107 ( Pt 12), 3569-3577
9 Saraste,A. and Pulkki,K. (2000) Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 45, 528537
10 Leist,M. and Jaattela,M. (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev.
Mol. Cell Biol. 2, 589-598
11 Hannon,G.J. and Beach,D. (1994) p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest.
Nature 371, 257-261
12 Reynisdottir,I. et al. (1995) Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to
TGF-beta. Genes Dev. 9, 1831-1845
13 Sakuraba,H. et al. (2004) Transforming growth factor-{beta} regulates susceptibility of epithelial apoptosis in
murine model of colitis. Ann. N. Y. Acad. Sci. 1029, 382-384
14 Vetuschi,A. et al. (2002) Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate
sodium-induced colitis in rats. Dig. Dis. Sci. 47, 1447-1457
15 Suzuki,A. et al. (2000) Fas/Fas ligand expression and characteristics of primed CD45RO+ T cells in the
inflamed mucosa of ulcerative colitis. Scand. J. Gastroenterol. 35, 1278-1283
16 Doering,J. et al. (2004) Induction of T lymphocyte apoptosis by sulphasalazine in patients with Crohn's disease.
Gut 53, 1632-1638
17 Souza,H.S. et al. (2005) Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease:
evidence of altered expression of FasL and perforin cytotoxic pathways. Int. J. Colorectal Dis. 20, 277-286
18 Strater,J. et al. (1997) CD95 (APO-1/Fas)-mediated apoptosis in colon epithelial cells: a possible role in
ulcerative colitis. Gastroenterology 113, 160-167
- 63 -
Chapter 2
19 Sturm,A. et al. (2004) Divergent cell cycle kinetics underlie the distinct functional capacity of mucosal T cells
in Crohn's disease and ulcerative colitis. Gut 53, 1624-1631
20 Yukawa,M. et al. (2002) Systemic and local evidence of increased Fas-mediated apoptosis in ulcerative colitis.
Int. J. Colorectal Dis. 17, 70-76
21 Hale,L.P. et al. (2005) Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and
mucosal exposure to intestinal bacteria. Inflamm. Bowel. Dis. 11, 1060-1069
22 Vousden,K.H. (2002) Activation of the p53 tumor suppressor protein. Biochim. Biophys. Acta 1602, 47-59
23 Kubbutat,M.H. et al. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299-303
24 Honda,R. and Yasuda,H. (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of
Mdm2 for tumor suppressor p53. EMBO J. 18, 22-27
25 Henriksson,M. et al. (2001) Inactivation of Myc-induced p53-dependent apoptosis in human tumors. Apoptosis.
6, 133-137
26 Krishna,M. et al. (1995) Expression of p53 antigen in inflamed and regenerated mucosa in ulcerative colitis and
Crohn's disease. Mod. Pathol. 8, 654-657
27 Scaffidi,C. et al. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17, 1675-1687
28 Sartorius,U. et al. (2001) Molecular mechanisms of death-receptor-mediated apoptosis. Chembiochem. 2, 20-29
29 Denault,J.B. and Salvesen,G.S. (2002) Caspases: keys in the ignition of cell death. Chem. Rev. 102, 4489-4500
30 Brannigan,A.E. et al. (2000) Neutrophil apoptosis is delayed in patients with inflammatory bowel disease.
Shock 13, 361-366
31 Irmler,M. et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388, 190-195
32 Thome,M. and Tschopp,J. (2001) Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev.
Immunol. 1, 50-58
33 Monteleone,I. et al. (2006) A functional role of flip in conferring resistance of Crohn's disease lamina propria
lymphocytes to FAS-mediated apoptosis. Gastroenterology 130, 389-397
34 Boirivant,M. et al. (1999) Lamina propria T cells in Crohn's disease and other gastrointestinal inflammation
show defective CD2 pathway-induced apoptosis. Gastroenterology 116, 557-565
35 Bregenholt,S. et al. (2001) The majority of lamina propria CD4(+) T-cells from scid mice with colitis undergo
Fas-mediated apoptosis in vivo. Immunol. Lett. 78, 7-12
36 Declercq,W. et al. (1998) Cooperation of both TNF receptors in inducing apoptosis: involvement of the TNF
receptor-associated factor binding domain of the TNF receptor 75. J. Immunol. 161, 390-399
37 Haridas,V. et al. (1998) Overexpression of the p80 TNF receptor leads to TNF-dependent apoptosis, nuclear
factor-kappa B activation, and c-Jun kinase activation. J. Immunol. 160, 3152-3162
38 Vandenabeele,P. et al. (1995) Both TNF receptors are required for TNF-mediated induction of apoptosis in
PC60 cells. J. Immunol. 154, 2904-2913
- 64 -
Apoptosis as a therapeutic paradigm in inflammatory bowel diseases
39 Weiss,T. et al. (1998) TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFRassociated factor 2 and is specific for TNFR60. J. Immunol. 161, 3136-3142
40 Holtmann,M.H. et al. (2002) Tumor necrosis factor-receptor 2 is up-regulated on lamina propria T cells in
Crohn's disease and promotes experimental colitis in vivo. Eur. J. Immunol. 32, 3142-3151
41 Antonsson,B. et al. (2000) Bax oligomerization is required for channel-forming activity in liposomes and to
trigger cytochrome c release from mitochondria. Biochemical Journal 345, 271-278
42 Tsujimoto,Y. and Shimizu,S. (2000) VDAC regulation by the Bcl-2 family of proteins. Cell Death. Differ. 7,
1174-1181
43 Reed,J.C. et al. (1998) Bcl-2 family proteins and mitochondria. Biochimica et Biophysica Acta-Bioenergetics
1366, 127-137
44 Krajewski,S. et al. (1994) Immunohistochemical determination of in vivo distribution of Bax, a dominant
inhibitor of Bcl-2. Am. J. Pathol. 145, 1323-1336
45 Ohman,L. et al. (2002) Regression of Peyer's patches in G alpha i2 deficient mice prior to colitis is associated
with reduced expression of Bcl-2 and increased apoptosis. Gut 51, 392-397
46 Zingarelli,B. et al. (2004) Activator protein-1 signalling pathway and apoptosis are modulated by poly(ADPribose) polymerase-1 in experimental colitis. Immunology 113, 509-517
47 Ina,K. et al. (1999) Resistance of Crohn's disease T cells to multiple apoptotic signals is associated with a Bcl2/Bax mucosal imbalance. J. Immunol. 163, 1081-1090
48 Itoh,J. et al. (2001) Decreased Bax expression by mucosal T cells favours resistance to apoptosis in Crohn's
disease. Gut 49, 35-41
49 Wahl,C. et al. (1998) Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest
101, 1163-1174
50 Tiede,I. et al. (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human
CD4+ T lymphocytes. J. Clin. Invest 111, 1133-1145
51 Elliott,M.J. et al. (1993) Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor
necrosis factor alpha. Arthritis Rheum. 36, 1681-1690
52 Moreland,L.W. et al. (1997) Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor
receptor (p75)-Fc fusion protein. N. Engl. J. Med. 337, 141-147
53 Van Den Brande,J.M. et al. (2003) Infliximab but not etanercept induces apoptosis in lamina propria Tlymphocytes from patients with Crohn's disease. Gastroenterology 124, 1774-1785
54 Ten Hove,T. et al. (2002) Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's
disease. Gut 50, 206-211
55 Atreya,R. et al. (2000) Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis
in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 6, 583588
56 Ito,H. et al. (2002) Anti-IL-6 receptor monoclonal antibody inhibits leukocyte recruitment and promotes T-cell
apoptosis in a murine model of Crohn's disease. J. Gastroenterol. 37 Suppl 14, 56-61
- 65 -
Chapter 2
57 Mudter,J. et al. (2002) A new model of chronic colitis in SCID mice induced by adoptive transfer of CD62L+
CD4+ T cells: insights into the regulatory role of interleukin-6 on apoptosis. Pathobiology 70, 170-176
58 Fuss,I.J. et al. (1999) Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in
mice. Gastroenterology 117, 1078-1088
59 Fichtner-Feigl,S. et al. (2005) Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with
NF-kappa B decoy oligonucleotides. J. Clin. Invest 115, 3057-3071
60 Xu,X.M. et al. (2005) Effects of garlicin on apoptosis in rat model of colitis. World J. Gastroenterol. 11, 45794582
61 Martin,A.R. et al. (2004) Resveratrol, a polyphenol found in grapes, suppresses oxidative damage and
stimulates apoptosis during early colonic inflammation in rats. Biochem. Pharmacol. 67, 1399-1410
62 Ohman,L. et al. (2005) Acellular Bordetella pertussis vaccine enhances mucosal interleukin-10 production,
induces apoptosis of activated Th1 cells and attenuates colitis in Galphai2-deficient mice. Clin. Exp. Immunol.
141, 37-46
- 66 -
Chapter
3.
Inhibition of soluble TNF- by single
domain camel antibodies does not
prevent experimental colitis
Marleen I. Verstege1, Fiebo J. ten Kate2, Anje A. te Velde1
1. Centre for Experimental and Molecular Medicine, Academic Medical Centre,
Amsterdam, The Netherlands
2. Department of Pathology, Academic Medical Centre, Amsterdam, The Netherlands
Chapter 3
Abstract
Tumour necrosis factor (TNF)- is a pro-inflammatory cytokine, which plays an important
role in the pathogenesis of inflammatory bowel diseases. Blocking of TNF- has been
demonstrated to be an effective strategy. However, most of the anti-TNF drugs have sideeffects and are immunogenic. In addition, there is differential binding to the two forms of
TNF-: the transmembrane (tmTNF-) and the soluble protein (sTNF-). In this study we
have tested a new inhibitor of sTNF- based on the heavy chains of camel antibodies, the
so-called nanobodies, in an acute and a chronic colitis mouse model. We demonstrated that
these nanobodies do not ameliorate colitis: there were no differences in body weight, colon
length, colon weight and histology between the treated and control group in both models.
Only in the acute model nanobodies decreased the amount of cells in the caudal lymph
nodes, but increased the cytokine production. The low efficacy of blocking of sTNF-
compared to a tmTNF- blocking antibody might reflect their role in the inflammatory
process.
- 68 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
Introduction
Crohn’s disease (CD) is characterised by chronic inflammation of the gastrointestinal tract
of which the pathogenesis is unknown. Several genetic, immunological and environmental
factors all contribute to the initiation and maintenance of the disease. It has been
demonstrated that an exaggerated immune response against the endogenous microflora by T
helper (Th) 1 and Th17 lymphocytes plays an important role in the pathogenesis of CD.
This immune response is characterised by an increase of pro-inflammatory cytokines,
including tumour necrosis factor (TNF)-, interleukin (IL)-1, transforming growth factor interferon- and IL-17 in the inflamed mucosa of CD patients. High concentrations of
TNF- can also be detected in the stool of CD patients
1,2
. That TNF- is a key player in
the pathogenesis of CD has been shown by overexpression of TNF- in mice, which results
in the development of chronic inflammatory arthritis and Crohn’s like IBD 3.
TNF- is first synthesised as a 26kDa transmembrane protein (tmTNF-) with an
intracellular tail. The metalloproteinase TNF- concerting enzyme (TACE) cleaves
tmTNF- into a soluble protein of 17kDa (sTNF-)
4,5
. Many cell types, also non-immune
cell types are able to produce TNF-, however the majority of TNF- is produced by
monocytes and macrophages 6. TNF- plays an important role in cell recruitment, cell
proliferation, apoptosis and immune regulation via their interaction with two different TNF receptors. Both sTNF- and tmTNF- are capable to bind the 55kDa TNF receptor
(TNFR)1 (CD120a) and the 75kDa TNFR2 (CD120b). However, sTNF- has a higher
7,8
affinity for TNFR1, whereas tmTNF- prefers to bind TNFR2
. Dependent on the
metabolic state of the cell, receptor-mediated effects of tmTNF- and sTNF- result in
apoptosis or nuclear factor (NF)-B activation. TNFR1 is constitutively expressed on most
cell types, whereas TNFR2 is mainly expressed on endothelial and haematopoietic cells,
although during active inflammation in IBD patients and mice colitis models also epithelial
cells express high levels of TNFR2 9. Similar to TNF-, soluble TNF- receptors are
released by proteolytic cleavage of their transmembrane form
10-12
. Both forms of soluble
- 69 -
Chapter 3
TNF- receptors are capable to function as a natural TNF- antagonist by neutralising
sTNF- 13.
Since TNF- seems to be a key player in a number of diseases like CD,
rheumatoid arthritis (RA), sarcoïdosis, and psoriasis, strategies to neutralise TNF- have
been developed. These strategies include infliximab (chimeric IgG anti-TNF antibody),
adalimumab (human IgG1 monoclonal anti-TNF antibody), certoluzimab (polyethylene Fab
fragment of anti-TNF) and etanercept (TNF- receptor 2 IgG1 invariant tail fusion protein).
Although 60 to 70% of the CD patients benefit from this anti-TNF strategies
14-16
, these
therapies have also many (severe) side-effects including immunoreactivity and can only be
administered intravenously or subcutaneously. Moreover, many patients do not respond,
loose responsiveness or become intolerant to the current anti-TNF therapies. When this is
the case, other anti-TNF-based drugs can be effective 17-21. Therefore, companies are still
developing alternatives to neutralise TNF-.
We have investigated a TNF- inhibitor that is developed by Ablynx. This antiTNF- is based on the discovery by the Vrije Universiteit Brussel in Belgium that
camelidae produce functional antibodies that only contain heavy chains
22,23
. The isolated
single variable domain (VHH) of heavy-chain antibodies still harbours the full antigen
binding-capacity. These so-called nanobodies have the advantage that they are oral
available and they should be less immunogenic because of their size
24-26
. We have tested
two different nanobodies in an acute colitis model (TNBS-induced colitis) and a chronic
colitis model (CD4+CD45RBhigh transfer colitis). Unfortunately, both nanobodies did not
prevent experimental colitis.
- 70 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
Materials and methods
Induction of experimental colitis
The Animal Studies Ethics Committee of the University of Amsterdam, The Netherlands,
validated all experiments. 7-10 week-old BALB/c and C.B-17 severe combined
immunodeficient (SCID) mice were obtained from Harlan Sprague-Dawley (Horst, The
Netherlands). During the experiments, the BALB/c mice were housed under standard
conditions, whereas the SCID mice were maintained in filter-top cages under specific
pathogen-free conditions in our animal facility. All the mice were allowed free access to
water and food. The different groups are mentioned in table 1.
CD4+CD45RBhigh transfer
Group
1
CD4+CD45RBhigh
vehicle intragastric
n
9
daily 100l TRIS buffer intragastric
Schedule
2
CD4+CD45RBhigh
nanobody 3F-3F
9
daily 50g 3F-3F intragastric in TRIS buffer
3
CD4+CD45RBhigh
vehicle i.p.
9
3x/week 100l PBS i.p.
4
CD4+CD45RBhigh
nanobody 3F-3F HSA21
7
3x/week 50g 3F-3F-HSA21 in PBS i.p.
5
CD4+CD45RBhigh
-mouse TNF- mAb
9
3x/week 50g anti-mouse TNF- mAb i.p.
6
CD4+CD45RBhigh
CD4+CD45RBlow
6
Day 0 and 7 1mg TNBS
vehicle intragastric
n
10
2
Day 0 and 7 1mg TNBS
nanobody 3F-3F
10
2x/day 100g 3F-3F intragastric in TRIS buffer
3
4
5
Day 0 and 7 1mg TNBS
Day 0 and 7 1mg TNBS
Day 0 and 7 1mg TNBS
vehicle i.p.
nanobody 3F-3F HSA21
-mouse TNF- mAb
10
10
10
2x/day 100l PBS i.p.
2x/day 100g 3F-3F-HSA21 in PBS i.p
at day 3,6, 9 50g anti-mouse TNF- mAb i.p.
TNBS colitis
Group
1
Schedule
2x/day 100l TRIS buffer intragastric
Table 1. Experimental design
Induction of CD4+CD45RBhigh transfer colitis
Chronic CD4+CD45RBhigh transfer colitis was induced as described previously 27,28. Briefly,
CD4+ splenocytes from BALB/c mice were isolated by red cell lysis and negative selection
by using rat-anti-mouse monoclonal antibodies (mAbs) against B220 (clone RA3-6B2),
- 71 -
Chapter 3
Mac-1 (clone M1/70) and CD8 (clone 53-6.7) (gift from Dr. R. Mebius, Free University
Medical Centre, Amsterdam, The Netherlands). Cells that were stained with mAbs were
removed in a magnetic field by using sheep-anti-rat immunoglobulin G-coated magnetic
beads (Dynal, Hamburg, Germany). Finally, the resulting CD4+ cells were stained with
cychrome-conjugated CD4 and fluorescein isothiocyanate-conjugated CD45RB mAbs (BD
Biosciences, San Diego, CA) so that these cell populations could be sorted by flow
cytometry (BD Biosciences, San Diego, CA). CD4+CD45RBhigh T populations were 95%
pure and 1-4x105 of these cells were transferred to C.B-17 SCID mice as a single
intraperitoneal (i.p.) injection to induce colitis. As a negative control group we used mice
that were transferred with both the CD4+CD45RBhigh T lymphocytes as well as the
CD4+CD45RBlow T lymphocytes, since the CD4+CD45RBlow population T cells protects the
mice from developing colitis.
Induction of TNBS colitis
TNBS colitis was induced as described previously 29. Briefly, at day 0 and day 7, 1.0 mg of
2,4,6-trinitrobenzene sulphonic acid (TNBS; Sigma Chemical Co., St. Louis, MO)
dissolved in 40% ethanol (Merck, Darmstadt, Germany) was administrated rectally using a
vinyl catheter positioned three cm from the anus. During this procedure, the mice were
anaesthetised with isoflurane (1-chloro-2,2,2-trifluoroethyl-isoflurane-difluoromethyl-ether;
Abbott Laboratories Ltd., Queenborough, Kent, England). After instillation, the mice were
kept vertically for 60 seconds.
Cell Culture and cytokine measurements
Caudal lymph node (CLN) cells of mice with TNBS-induced colitis were isolated by
passing the lymph node through a 40m filter cell strainer (Becton/Dickson Labware, New
Jersey, USA). The isolated lymphocytes were suspended in 4 ml RPMI 1640 containing Lglutamine, 10% foetal calf’s serum and antibiotics (penicillin G sodium 10000 U/ml,
streptomycin sulphate 25 g/ml, amphotericin B 25 g/ml) (all from Gibco/BRL, Paisley,
Scotland). The cells were counted and added to flat-bottom 96-well plates at 2 x 105 cells
- 72 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
per well in a total volume of 200l of the same medium. The cells were cultured in the
presence of immobilised -CD3 (1:30; 145.2C11 clone) and soluble -CD28 (1:1000;
PharMingen, San Diego, CA) for 48 hours at 37°C. The supernatant was collected and used
for a cytometric bead array (CBA) (BD Biosciences, San Jose, CA). A CBA was performed
to determine simultaneously the production of TNF-, IFN-, IL-2, IL-4 and IL-5
according to the manufactures recommendations. Two-colour flow cytometric analysis was
performed using FACScan® flow cytometre (Becton Dickonson Immunocytometry
Systems (BDIS), San Jose, CA). Data were acquired and analysed using Becton Dickinson
CBA software.
Parameters to assess inflammation
The weight of the mice was recorded every day (TNBS-colitis) or twice a week
(CD4+CD45RBhigh colitis). Mice had to be sacrificed if their weight loss is more than 15%
compared to their initial weight or more than 25% compared to the control group without
colitis. After sacrificing the mice, the CLN and the colon were collected. Through a midline
incision, the colons were removed. Length and after removing the faecal material, weight of
the colons (calculated for 6 cm) were measured and used as an indicator of disease-related
intestinal shortening and thickening, respectively.
Histological examination
Longitudinally divided rolled-up parts of colons were fixed in 4% buffered formalin in PBS
for 24 hours and embedded in paraffin for routine histology. Three transverse slices (5 m),
taken from each colonic sample, were stained with haematoxylin-eosine and examined by
light microscopy. Colonic inflammation was evaluated microscopically in a blinded manner
by an experienced pathologist. Inflammation induced by TNBS-colitis was estimated by 1)
percentage of involved area, 2) the amount of follicles, 3) oedema, 4) fibrosis, 5)
erosion/ulceration, 6) crypt loss and infiltration of 7) granulocytes and 8) monocytes with a
maximum score of 24. Inflammation induced by CD4+CD45RBhigh colitis was estimated by
the 1) percentage of involved area, 2) depletion of goblet cells, 3) epithelial hyperplasia, 4)
- 73 -
Chapter 3
ulcerations, 5) crypt abscesses and 6) infiltration of granulocytes and monocytes with a
maximal score of 4.
Statistical analysis
All data are expressed as the means ± the standard error of the mean (SEM) and were
analysed using Graphpad prism 4 (Graphpad Prism v. 4 for Windows, GraphPad Software,
San Diego, California USA). Differences between groups were analysed using the nonparametric Mann Whitney U test. Changes in body weight between the four groups were
analysed by one-way ANOVA with a Bonferonni post-hoc test when differences between
interventions were significantly different. Correlation analyses were performed using the
Spearman correlation test. All statistics were performed two-tailed and values of p<0.05
were considered statistically significant (* p<0.05; ** p<0.01, *** p<0.001).
- 74 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
Results
Nanobodies do not ameliorate CD4+CD45RBhigh transfer colitis
SCID mice that have received CD4+CD45RBhigh T lymphocytes (called CD4+CD45RBhigh
transferred mice) will develop colitis in approximately four weeks characterised by wasting
disease, diarrhoea and inflammation. Colitis induced by the transfer of CD4+CD45RBhigh T
lymphocytes is accompanied by the loss of body weight (see figure 1.). All mice survived
this induction of colitis, but body weight changes were significantly different between the
six groups (p<0.0001). CD4+CD45RBhigh transferred mice treated with nanobody 3F-3F
significantly developed more wasting disease compared to CD4+CD45RBhigh transferred
mice
+
that
were
high
CD4 CD45RB
treated
+
with
low
CD4 CD45RB
anti-mouse
TNF-
(p<0.01)
and
the
+
transferred mice (p<0.001). Also CD4 CD45RBhigh
transferred mice that received 3F-3F HSA21 developed more wasting disease compared to
CD4+CD45RBhigh transferred mice (p<0.01), anti-mouse TNF--treated CD4+CD45RBhigh
transferred mice (p<0.001) and CD4+CD45RBhighCD4+CD45RBlow transferred mice
(p<0.001). Moreover, at the day of sacrifice CD4+CD45RBhigh transferred mice treated with
nanobody 3F-3F or 3F-3F HSA21 have lost significantly more weight than
CD4+CD45RBhigh transferred mice treated with anti-mouse TNF- mAb (p=0.008 and
p=0.01, respectively) and CD4+CD45RBhighCD4+CD45RBlow transferred mice (p=0.0008
and p=0.0007, respectively).
Under influence of inflammation, the muscles of the colon contract resulting in a
shortening of the colon and an influx of inflammatory cells and oedema results in an
increased weight of the colon. After sacrificing the mice, length and weight of the last six
centimetres of the colon were determined. Although body weight changes correlated with
colon length (r=0.421, p=0.002), there were no statistically significant differences in colon
length between the different groups (see figure 2). Weight of the colon is negatively
correlated with body weight changes (r=-0.503, p=0.0002) and with colon length (r=-0.403,
p=0.0041), indicating that an increase of colon weight is associated with a loss of body
weight and a decrease in colon length. The weight of the colon is significantly increased in
- 75 -
Chapter 3
CD4+CD45RBhigh transferred mice that were treated with the 3F-3F nanobody (347.9 ±
38.9mg) compared to CD4+CD45RBhigh transferred mice treated with anti-mouse TNFmAb (214.3 ± 19.2mg; p=0.006) and CD4+CD45RBhighCD4+CD45RBlow transferred mice
(212.2 ± 15.5mg; p=0.005). CD4+CD45RBhigh transferred mice that received the 3F-3F
nanobody had significant increased colon weight compared to CD4+CD45RBhigh transferred
mice treated with intragastric vehicle (347.9 ± 38.9mg vs. 245.3 ± 20.4;.p=0.02). There
were no significant differences in colon weight between 3F-3F HSA-21 treated
CD4+CD45RBhigh transferred mice and CD4+CD45RBhigh transferred mice treated with
vehicle i.p. (305.7 ± 37.2mg vs. 268.1 ± 11.5mg; p=0.6, respectively). These results
indicate that both nanobodies are not able to reduce colitis in the CD4+CD45RBhigh transfer
model since colon weight is not decreased, compared to CD4+CD45RBhigh transferred mice
treated with vehicle.
Colonic inflammation was also analysed by histological analyses (see figure 3.).
Histological scores of both the CD4+CD45RBhigh intragastric vehicle group (2.1 ± 0.2) and
the CD4+CD45RBhigh i.p. vehicle group (2.0 ± 0.2) were increased compared to the
CD4+CD45RBhighCD4+CD45RBlow transferred mice (1.2 ± 0.2; p=0.01 and p=0.02,
respectively) and the CD4+CD45RBhigh transferred mice treated with anti-mouse TNF-
(1.3 ± 0.2; p=0.02 and p=0.03, respectively), so transfer of CD4+CD45RBlow T lymphocytes
and treatment of -mouse TNF- ameliorates colitis in CD4+CD45RBhigh transferred mice.
There were no statistically significant differences in histological score between the
CD4+CD45RBhigh intragastric vehicle group and the 3F-3F nanobody-treated group (2.1 ±
0.2 vs. 1.7 ± 0.2; p=0.2) and the 3F-3F HSA21 nanobody-treated group compared to the i.p.
vehicle group (1.7 ± 0.2 vs.2.0 ± 0.2; p=0.4), indicating that nanobody treatment does not
result in a decrease of histological parameters. Body weight changes were negatively
correlated with histological scores (r=-.0336, p=0.02), indicating that loss of body weight is
associated with an increase in histological scores.
- 76 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
120
***
110
*
***
100
**
125
80
100
Weight loss (%)
90
70
80
Day
CD4+ CD45RBhigh intragastric
CD4+ CD45RBhigh + 3F-3F
CD4+ CD45RBhigh i.p.
CD4+ CD45RBhigh + 3F-3F-HSA21
CD4+ CD45RBhigh+ anti TNF mAb
CD4+ CD45RBhighCD4+ CD45RBlow
50
25
w
0
hi
gh
B
45
R
D
+
C
C
D
D
4
C
C
a.
75
ig
ic
C
h
D
4+
+
D
3F
C
45
D
-3
4
R
4+
5R
F
Bh
C
ig
Bh
D
h
ig
45
h
+
R
C
3F
i.p
Bh
D
ig
-3
.
4+
h
FC
+
H
D
an
45
SA
R
ti
21
Bh
T
ig
N
h
F
C
D
m
4+
A
b
C
D
45
R
B lo
70
+
60
C
50
D
4
40
b.
C
D
30
in
tr
ag
45
as
R
Bh
tr
20
+
10
C
D
0
4
Weight loss (%)
130
*
*
**
**
*
*
11
300
1.0
100
b
45
R
B
lo
w
21
A
m
C
D4
+
C
D
-H
NF
D
45
R
B
hi
gh
+
hi
gh
an
t
iT
-3
F
3F
+
RB
C
+
D
4
C
+
D4
SA
i.p
.
hi
gh
3F
-
+
5R
B
D4
+
C
hi
gh
D4
45
RB
D
C
C
D
45
C
D
e.
C
in
t
h
hig
45
R
B
D
5R
+
C
D4
D4
C
+
C
D
4
C
hi
gh
ra
g
as
tr
ic
lo
w
ra
ga
st
ric
D
C
4+
D
4+
C
+
D
3F
C
C
45
D4
D4
3F
RB
+
5R
hi
C
gh
B hi
D4
gh
5R
+
C
3
i
B
.p
hi
FD
gh
4+
.
3F
C
-H
+
D
an
SA
45
R
21
B h i ti T
N
gh
F
C
D4
m
+
A
b
C
D
45
RB
hi
gh
in
t
h
hig
45
R
B
D
+
C
45
R
D
D4
+
C
C
+
C
4
D
+
C
D
C
4
D4
C
D
C
d.
3F
0.0
B
w
D
4+
+
D
3F
C
45
D
-3
45
RB
F
hig
R
B hi
h
45
gh
+
R
C
i.p
B hig 3F
D4
-3
.
+
h
FC
+
H
D4
an
SA
5R
21
B hi ti T
NF
gh
C
D
m
4+
A
b
C
D
45
RB
lo
st
ric
ra
ga
hi
gh
in
t
RB
+
C
C
D4
5
hi
gh
R
B
D4
5
D4
+
C
C
D4
C
c.
0.5
0
7
+
8
1.5
200
4
9
2.0
B
10
2.5
C
Weight (mg)
400
C
Length (cm)
*
12
Figure 1. a. Bodyweight was recorded twice a week and weight changes were significant different between the
six groups (p<0.0001). b. At the day of sacrifice CD4+CD45RBhigh transferred mice treated with nanobody 3F-3F
or 3F-3F HSA21 lost significantly more weight than CD4+CD45RBhigh transferred mice treated with anti-mouse
TNF- mAb (p=0.008 and p=0.01, respectively) and CD4+CD45RBhighCD4+CD45RBlow transferred mice
(p=0.0008 and p=0.0007, respectively). c. Colon length was not significantly different between the six groups. d.
Colon weight was significantly increased in CD4+CD45RBhigh transferred mice treated with 3F-3F nanobody (■)
compared to CD4+CD45RBhigh transferred mice treated with vehicle (■) (p=0.02), CD4+CD45RBhigh transferred
mice treated with anti-mouse TNF- (■) (p=0.006) and CD4+CD45RBhighCD4+CD45RBlow transferred mice (■)
(p=0.005). e. Administration of soluble TNF- inhibitors did not prevent inflammation of the colon. There are no
significant differences in histological scores between CD4+CD45RBhigh mice treated with nanobody 3F-3F or 3F3F HSA21 and the other groups. However, histological scores of both the CD4+CD45RBhigh intragastric vehicle
group (■) (2.1 ± 0.2) and the CD4+CD45RBhigh i.p. vehicle group (■) (2.0 ± 0.2) were increased compared to the
CD4+CD45RBhighCD4+CD45RBlow transferred mice (■) (1.2 ± 0.2; p=0.01 and p=0.02, respectively) and the
CD4+CD45RBhigh transferred mice treated with anti-mouse TNF-(■) (1.3 ± 0.2; p=0.02 and p=0.03,
respectively).
- 77 -
Chapter 3
Nanobodies do not prevent TNBS-induced colitis
To determine the effect of nanobodies on the development of TNBS colitis, the mice
received 1mg TNBS intrarectally at day 0 and day 7, and after 9 days the mice were
sacrificed. Because of the second administration of TNBS, a delayed type hypersensitivity
reaction will occur and the mice develop wasting disease, diarrhoea and inflammation of
the colon. Mice were treated with 3F-3F orally or i.p. The weight of the mice was recorded
every day or every two days and after sacrificing the mice length and weight of the mice
were measured. Bodyweight loss was not significant different between the five groups
during the development of colitis and at the day of sacrifice (see figure 2.). Moreover, there
are no significant differences in colon length. The weight of the colon however is
significantly increased in mice treated with 3F-3F compared to mice that were treated with
anti-mouse TNF- (183.3 ± 7.4mg vs. 153.1 ± 4.4mg; p<0.001). The histological score of
the 3F-3F-treated group is also significantly higher compared to the vehicle-treated group
(9.8 ± 0.7 vs. 4.2 ± 0.9; p<0.0001). Besides, mice that were treated with anti-mouse TNFhave an increased histological score compared to the control mice (8.2 ± 0.6 vs. 4.2 ± 0.9;
p=0.002).
- 78 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
Weight loss (%)
110
105
100
95
7
8
9
10
Day
Vehicle intragastric
3F-3F
Vehicle i.p.
3F-3F HSA21
anti TNF- mAb
100
95
b
A
21
m
F
TN
tr
in
tr
ag
as
**
b
TN
F-
m
A
.
i.p
21
an
ti
tr
ag
a
H
SA
st
ric
A
b
m
TN
F-
in
.
21
i.p
3F
cl
e
3F
-
H
SA
3F
-3
F
an
ti
e.
Ve
hi
cl
e
d.
Ve
hi
in
tr
ag
as
tr
ic
A
b
m
F
an
ti
TN
i.p
.
cl
e
H
SA
21
3F
-3
F
Ve
hi
3F
-3
F
100
3F
125
3F
-
150
Ve
hi
cl
e
175
***
11
10
9
8
7
6
5
4
3
2
1
0
-3
F
Histology score
Weight (mg)
200
Ve
hi
cl
e
c.
Ve
hi
cl
e
in
tr
ag
as
tr
ic
Length (cm)
**
11
10
9
8
7
6
5
4
3
2
1
0
3F
b.
an
ti
ic
90
Ve
hi
cl
e
a.
105
i.p
.
6
le
5
HS
A
4
F
3
Ve
hi
c
2
3F
-3
1
3F
-3
F
0
Weight loss (%)
110
90
Figure 2. At day 0 and day 7, the mice received 1mg TNBS rectally (arrows in figure a.) and body weight loss
was recorded daily or every two days. Body weight loss during the time of the experiment (a.), weight loss at the
day of sacrifice (b.) and length of the colon (c.) were not significantly different between the five groups. d. Colon
weight was significantly increased in mice treated with 3F-3F compared to mice that were treated with antimouse TNF- (p=0.001). e. The histological score was significant increased in mice treated with 3F-3F compared
to mice that were treated with vehicle (p<0.0001). Also mice that were treated with anti-mouse TNF- have an
increased histological score compared to the control mice (p=0.002).
- 79 -
Chapter 3
Nanobodies decrease the amount of caudal lymph node lymphocytes, but
increase cytokine production
To determine the effect of nanobodies on cytokine production, CLNs were collected after
sacrificing the mice and the number of lymphocytes was counted. Thereafter, lymphocytes
were stimulated with anti-CD3 and anti-CD28 and supernatant was collected after 24 hours
to investigate the IFN- and TNF- production (see figure 3). Oral administration of 3F-3F
nanobody results in a significant decrease of the amount of lymphocytes in the CLN
compared to the control mice (2.0 x 105 vs. 4.2 x 105, p=0.02) and mice treated with antiTNF- mAb (2.0 x 105 vs. 4.1 x 105, p=0.04). Also mice treated with 3F-3F HSA21
showed a decrease in lymphocyte amount compared to the control mice and anti-TNF-
c.
***
**
1000
750
500
250
b
21
m
A
N
F-
ti
T
an
i.p
.
H
SA
cl
e
3F
-3
F
Ve
hi
3F
-3
F
0
in
tr
ag
as
tr
ic
IFN- production (pg/ml)
1250
Ve
hi
cl
e
b
21
m
A
TN
F
an
ti
H
SA
i.p
.
***
3F
-3
F
in
tr
hi
cl
e
Ve
b.
***
3F
-3
F
TNF- production (pg/ml)
m
ag
as
tr
ic
A
b
900
800
700
600
500
400
300
200
100
0
TN
F-
an
ti
i.p
.
H
SA
21
3F
-3
F
3F
-3
F
Ve
hi
cl
e
in
tr
ag
cl
e
Ve
hi
a.
*
Ve
hi
cl
e
*
5.5×10 5
5.0×10 5
4.5×10 5
4.0×10 5
3.5×10 5
3.0×10 5
2.5×10 5
2.0×10 5
1.5×10 5
1.0×10 5
5.0×10 4
0
as
tr
ic
Number of lymphocytes
mAb-treated mice, but these data were not significantly different.
Figure 3. Lymphocytes were isolated from the caudal lymph node and counted (a.). Thereafter these
lymphocytes were stimulated with anti-CD3 and anti-CD28 for 24 h and supernatant was collected to measure
TNF-(b.) and IFN- levels (c.).
Although mice treated with 3F-3F have significantly decreased lymphocyte
amounts, these lymphocytes produce significantly higher levels of TNF- and IFN-
compared to the control mice (678.8 ± 125.9 pg/ml vs. 82.6 ± 38.8pg/ml, p<0.0001 and
960.0 ± 240.9pg/ml vs. 18.5 ± 12.9 pg/ml, p<0.0001) and the anti-TNF- mAb-treated
mice (678.8 ± 125.9pg/ml vs. 117.6 ± 34.9pg/ml, p=0.0006 and 960.0 ± 240.9pg/ml vs.
148.7 ± 119.4pg/ml, p<0.004). The production of TNF- and IFN- by lymphocytes
isolated from mice treated with 3F-3F HSA21 is also increased, but is not significantly
different compared to the control group and the anti-TNF- mAb-treated group.
- 80 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
Discussion
Although TNF inhibitors such as infliximab and adalimumab are often used in the treatment
of CD, these drugs have many side-effects and can only be administered intravenously or
subcutaneously. Therefore pharmaceutical companies still develop alternatives to reduce
levels of TNF- in patients with CD and other inflammatory disorders like RA. We have
tested two types of nanobodies in a chronic colitis model (CD4+CD45RBhigh transferinduced colitis) and an acute colitis model (TNBS-induced colitis). Both nanobodies did not
ameliorate CD4+CD45RBhigh transfer colitis and TNBS-induced colitis. TNF- is present in
two forms, namely as a transmembrane and a soluble protein. Since these nanobodies block
the soluble form of TNF-only and do not decrease inflammatory processes, it seems that
sTNF- is necessary to regulate anti-inflammatory effects and/or that tmTNF- plays an
important role in pro-inflammatory processes. Probably not the soluble form of TNF- has
to be blocked, but the transmembrane form.
It has been shown earlier that infliximab and adalimumab are able to induce
apoptosis of T lymphocytes and monocytes, but that the soluble TNF receptor antagonist
etanercept is not
30-34
. Moreover, in contrast to RA patients, IBD patients do not have
clinical benefit of etanercept indicating that in IBD simply neutralising TNF- is not
enough to reduce inflammation 35. In addition, complete inhibition of TNF- secretion in
tmTNF transgenic RAG-/- mice could not prevent or delay colitis by transfer of
CD4+CD45high cells, although tmTNF- seems not to be important in the induction of
colitis since these mice also develop colitis if they receive TNF-deficient CD4+CD45high
cells 36.
The soluble form of TNF- has a higher affinity for binding to TNFR1 compared
to tmTNF- 8. It has been shown in mice that are NEMO deficient in intestinal epithelial
cells, that they are more sensitive to colitis, since NEMO deficiency activates TNF-induced apoptosis, whereas the inactivation of TNFR1 leads to inhibition of intestinal
inflammation
37
. In contrast to TNFR2, TNFR1 contains a cytoplasmic death domain and
- 81 -
Chapter 3
although in general activation of TNFR1 results in the induction of the NF-B pathway,
when a viral infection modifies the metabolic state of the cell the apoptotic pathway
through caspase activation is initiated
38
. Blocking of sTNF- may lead to an impaired
apoptosis in IBD patients resulting in survival of reactive T cells which can maintenance
inflammatory processes. Moreover, tmTNF- has a higher affinity for TNFR2 and seems to
be more involved in cell survival processes instead of cell death
7,39
. Blocking of only
sTNF- may result in an increased activation of TNFR2 by tmTNF- and consequently in
an increase of cell survival of reactive T lymphocytes.
Interestingly, tmTNF- functions not merely as a ligand for TNFRs, but it can also
acts as a receptor since binding to TNFRs and TNF antagonisten may induce reverse
signalling and consequently induce cell activation, cytokine suppression, or apoptosis of the
tmTNF- bearing cell
40-44,44
. Although both etanercept and infliximab bind to tmTNF-,
only infliximab is able to induce reverse signalling resulting in apoptosis, IL-10 and TGF-
production and cell cycle G0/G1 arrest
34,45
. Typically, serum levels of sTNFR1 and
sTNFR2 are increased in IBD patients compared to healthy controls and especially sTNFR1
is upregulated in serum of CD patients 46, indicating that these increased sTNFR levels are
a feedback mechanism to endeavour reduction of inflammation, but fail because of a
defective pathway in these patients. This hypothesis is supported by the fact that a lack of
TNFR2 expression by CD4+ lymphocytes results in an exacerbation of experimental colitis
47
. However, another study showed that also overexpression of TNFR2 in CD4+
lymphocytes results in an exacerbation of experimental colitis, probably through an
enhanced NF-B activation via the membrane form of TNFR2
48
. Moreover, infliximab
decreases the expression of tmTNFR2 on monocytes, accordingly reducing the action of
TNF-, but increases the secretion TNFR2 by monocytes, thereby contributing to their
neutralising capacity 49. Taken together, these results and our data demonstrate that TNF-
and its receptors may have different functions during inflammation dependent on the
metabolic state of the cell and the microenvironment and that a better understanding of
specific characteristics of TNF signalling will be the basis for the development of more
efficient therapeutics.
- 82 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
In conclusion, soluble TNF inhibitors seem not to be effective in experimental
colitis, probably because sTNF- has anti-inflammatory capacities and/ or tmTNF-
functions as pro-inflammatory ligand, which can be blocked by reverse signalling. However,
more research is necessary to investigate how sTNF- and tmTNF- acts during
inflammation and whether tmTNF- inhibitors are more effective in the treatment of IBD
than sTNF- inhibitors.
- 83 -
Chapter 3
References
1 Braegger,C.P. et al. (1992) Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet
339, 89-91
2 Nicholls,S. et al. (1993) Cytokines in stools of children with inflammatory bowel disease or infective diarrhoea.
J. Clin. Pathol. 46, 757-760
3 Kontoyiannis,D. et al. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich
elements: implications for joint and gut-associated immunopathologies. Immunity. 10, 387-398
4 Black,R.A. et al. (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells.
Nature 385, 729-733
5 Moss,M.L. et al. (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis
factor-alpha. Nature 385, 733-736
6 Vassalli,P. (1992) The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 10, 411-452
7 Grell,M. et al. (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80
kDa tumor necrosis factor receptor. Cell 83, 793-802
8 Ksontini,R. et al. (1998) Revisiting the role of tumor necrosis factor alpha and the response to surgical injury
and inflammation. Arch. Surg. 133, 558-567
9 Holtmann,M.H. et al. (2002) Tumor necrosis factor-receptor 2 is up-regulated on lamina propria T cells in
Crohn's disease and promotes experimental colitis in vivo. Eur. J. Immunol. 32, 3142-3151
10 Bjornberg,F. et al. (1994) Mechanisms involved in the processing of the p55 and the p75 tumor necrosis factor
(TNF) receptors to soluble receptor forms. Lymphokine Cytokine Res. 13, 203-211
11 Hawari,F.I. et al. (2004) Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: a mechanism
for generation of soluble cytokine receptors. Proc. Natl. Acad. Sci. U. S. A 101, 1297-1302
12 Porteu,F. and Nathan,C. (1990) Shedding of tumor necrosis factor receptors by activated human neutrophils. J.
Exp. Med. 172, 599-607
13 Pinckard,J.K. et al. (1997) Constitutive shedding of both p55 and p75 murine TNF receptors in vivo. J.
Immunol. 158, 3869-3873
14 Louis,E. et al. (2002) A positive response to infliximab in Crohn disease: association with a higher systemic
inflammation before treatment but not with -308 TNF gene polymorphism. Scand. J. Gastroenterol. 37, 818-824
15 Vermeire,S. et al. (2005) The role of C-reactive protein as an inflammatory marker in gastrointestinal diseases.
Nat. Clin. Pract. Gastroenterol. Hepatol. 2, 580-586
16 Vermeire,S. et al. (2006) Laboratory markers in IBD: useful, magic, or unnecessary toys? Gut 55, 426-431
17 Barthel,H.R. et al. (2005) Successful treatment with adalimumab in infliximab-resistant Crohn's disease. J.
Gastroenterol. Hepatol. 20, 1464-1465
- 84 -
Inhibition of sTNF- by single domain camel antibodies does not prevent experimental colitis
18 Gomez-Reino,J.J. and Carmona,L. (2006) Switching TNF antagonists in patients with chronic arthritis: an
observational study of 488 patients over a four-year period. Arthritis Res. Ther. 8, R29
19 Nikas,S.N. et al. (2006) Efficacy and safety of switching from infliximab to adalimumab: a comparative
controlled study. Ann. Rheum. Dis. 65, 257-260
20 Sandborn,W.J. et al. (2004) An open-label study of the human anti-TNF monoclonal antibody adalimumab in
subjects with prior loss of response or intolerance to infliximab for Crohn's disease. Am. J. Gastroenterol. 99,
1984-1989
21 van Vollenhoven,R. et al. (2003) Treatment with infliximab (Remicade) when etanercept (Enbrel) has failed or
vice versa: data from the STURE registry showing that switching tumour necrosis factor alpha blockers can make
sense. Ann. Rheum. Dis. 62, 1195-1198
22 Muyldermans,S. (2001) Single domain camel antibodies: current status. J. Biotechnol. 74, 277-302
23 Wolfson,W. (2006) Ablynx makes nanobodies from llama bodies. Chem. Biol. 13, 1243-1244
24 Cortez-Retamozo,V. et al. (2002) Efficient tumor targeting by single-domain antibody fragments of camels. Int.
J. Cancer 98, 456-462
25 Cortez-Retamozo,V. et al. (2004) Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 64,
2853-2857
26 Verheesen,P. et al. (2006) Prevention of oculopharyngeal muscular dystrophy-associated aggregation of
nuclear polyA-binding protein with a single-domain intracellular antibody. Hum. Mol. Genet. 15, 105-111
27 Morrissey,P.J. et al. (1993) CD4+ T cells that express high levels of CD45RB induce wasting disease when
transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer
of purified CD4+ T cells. J. Exp. Med. 178, 237-244
28 Powrie,F. et al. (1993) Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal
inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461-1471
29 Ten,H.T. et al. (2001) Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local
TNF-alpha production in mice. Gastroenterology 121, 1372-1379
30 Mitoma,H. et al. (2008) Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on
transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and
adalimumab. Arthritis Rheum. 58, 1248-1257
31 Shen,C. et al. (2005) Adalimumab induces apoptosis of human monocytes: a comparative study with infliximab
and etanercept. Aliment. Pharmacol. Ther. 21, 251-258
32 Ten Hove,T. et al. (2002) Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's
disease. Gut 50, 206-211
33 van den Brande,J.M. et al. (2003) Infliximab but not etanercept induces apoptosis in lamina propria Tlymphocytes from patients with Crohn's disease. Gastroenterology 124, 1774-1785
34 Waetzig,G.H. et al. (2005) Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF
signaling and autocrine transforming growth factor-beta1. FASEB J. 19, 91-93
- 85 -
Chapter 3
35 Sandborn,W.J. et al. (2001) Etanercept for active Crohn's disease: a randomized, double-blind, placebocontrolled trial. Gastroenterology 121, 1088-1094
36 Corazza,N. et al. (2004) Transmembrane tumor necrosis factor is a potent inducer of colitis even in the absence
of its secreted form. Gastroenterology 127, 816-825
37 Nenci,A. et al. (2007) Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446,
557-561
38 Ware,C.F. (2005) Network communications: lymphotoxins, LIGHT, and TNF. Annu. Rev. Immunol. 23, 787819
39 Wallach,D. et al. (1997) Cell death induction by receptors of the TNF family: towards a molecular
understanding. FEBS Lett. 410, 96-106
40 Eissner,G. et al. (2000) Reverse signaling through transmembrane TNF confers resistance to
lipopolysaccharide in human monocytes and macrophages. J. Immunol. 164, 6193-6198
41 Eissner,G. et al. (2004) Ligands working as receptors: reverse signaling by members of the TNF superfamily
enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 15, 353-366
42 Harashima,S. et al. (2001) Outside-to-inside signal through the membrane TNF-alpha induces E-selectin
(CD62E) expression on activated human CD4+ T cells. J. Immunol. 166, 130-136
43 Kirchner,S. et al. (2004) Effect of different tumor necrosis factor (TNF) reactive agents on reverse signaling of
membrane integrated TNF in monocytes. Cytokine 28, 67-74
44 Watts,A.D. et al. (1999) Soluble TNF-alpha receptors bind and neutralize over-expressed transmembrane TNFalpha on macrophages, but do not inhibit its processing. J. Leukoc. Biol. 66, 1005-1013
45 Mitoma,H. et al. (2005) Infliximab induces potent anti-inflammatory responses by outside-to-inside signals
through transmembrane TNF-alpha. Gastroenterology 128, 376-392
46 Spoettl,T. et al. (2007) Serum soluble TNF receptor I and II levels correlate with disease activity in IBD
patients. Inflamm. Bowel. Dis. 13, 727-732
47 Dayer,S.J. et al. (2009) Lack of TNFR2 expression by CD4(+) T cells exacerbates experimental colitis. Eur. J.
Immunol.
48 Holtmann,M.H. et al. (2002) Tumor necrosis factor-receptor 2 is up-regulated on lamina propria T cells in
Crohn's disease and promotes experimental colitis in vivo. Eur. J. Immunol. 32, 3142-3151
49 Ebert,E.C. (2009) Infliximab and the TNF-alpha system. Am. J. Physiol Gastrointest. Liver Physiol 296, G612G620
- 86 -
Part 2
Dendritic cell populations
and C-type lectins in
inflammatory bowel
diseases
Chapter
4.
Dendritic cell populations in colon and
mesenteric lymph nodes of patients with
Crohn’s disease
Marleen I. Verstege1, Fiebo J.W. ten Kate2, Susanne M. Reinartz 3, Cornelis M. van
Drunen3, Frederik J.M. Slors4, Willem A. Bemelman4, Florry A. Vyth-Dreese5, Anje
A. te Velde1
1. Centre for Experimental and Molecular Medicine, Academic Medical Centre,
Amsterdam, The Netherlands
2. Department of Pathology, Academic Medical Centre, Amsterdam, The Netherlands
3. Department of Otorhinolaryngology, Academic Medical Centre, Amsterdam, The
Netherlands
4. Department of Surgery, Academic Medical Centre, Amsterdam, The Netherlands
5. Department of Immunology, NKI-AVL, Amsterdam, The Netherlands
Chapter 4
Abstract
Dendritic cells (DCs) are key cells in innate and adaptive immune responses that determine
the pathophysiology of Crohn's disease. Intestinal DCs migrate from the mucosa into
mesenteric lymph nodes (MLNs). A number of different markers are described to define the
DC populations. In this study we have identified the phenotype and localisation of intestinal
and MLN DCs in patients with Crohn's disease and non-IBD patients based on these
markers. We used immunohistochemistry to demonstrate that all markers (s-100, CD83,
DC-SIGN, BDCA1-4, and CD1a) showed a different staining pattern varying from
localisation in T-cell areas of lymph follicles around blood vessels or single cells in the
lamina propria and in the MLN in the medullary cords and in the subcapsular sinuses
around blood vessels and in the T-cell areas. In conclusion, all different DC markers give
variable staining patterns so there is no marker for the DC.
- 90 -
DC populations in colon and MLNs of patients with CD
Introduction
Inflammatory bowel diseases (IBD) are chronic inflammatory diseases of the gut leading to
Crohn's disease (CD) or ulcerative colitis (UC). The pathogenesis of these diseases is not
well understood, but evidence is increasing that dendritic cells (DCs) play an important role
in the induction and maintenance of chronic inflammation
1,2
. DCs of CD patients seem to
have an intrinsic abnormal responsiveness to antigens from the lumen of the gut. Mutations
in receptors and/or signal transduction molecules may cause altered recognition of antigens
such as NOD2 mutations 3-6. However, it is not yet known what DC populations are present
in inflamed and control colon and mesenteric lymph nodes (MLNs).
For characterisation of human DCs, a series of markers have been used. In
peripheral blood, five distinct subsets of DCs have been identified (see table 17-18). In
addition, myeloid and plasmacytoid DCs can be distinguished (see table 17-18). Baumgart et
al. demonstrated that, in blood of IBD patients during flare-ups of the disease, immature
DCs of both myeloid and plasmacytoid origins are reduced, probably because these cells
migrate to the gut 19.
Blood DCs
subsets
references
CD1b/CD1c
Myeloid
CD11c+
CD16
BDCA3
Plasmacytoid
Stem cell
CD11c-
8,9,12,14
CD123/ BDCA2/ BDCA4
CD34
subsets
references
langerin/ CD1a/ s-100
7, 10, 16, 17
immature DCs
CD209
11
mature DCs
CD83
18
CD123/ BDCA2/ BDCA4
8, 9, 12
Tissue DCs
Langerhans cells
Myeloid
Plasmacytoid
dermal/ tissue/ interstitial DCs
Table 1. Markers that can be used for the characterisation of DC populations in blood and tissue.
- 91 -
Chapter 4
In tissues, three major human DC populations are distinguished, i.e. two myeloidderived DC populations and one plasmacytoid DC population. Table 2 lists the
characteristics of the different DC populations in peripheral tissues 7-9,11,16,18,20-24.
In the present study we have determined which DC subpopulations in human
colon and MLN can be distinguished when these different markers are used. In addition, we
speculate which of these populations may be involved in the pathogenesis of CD. As far as
we know, we have performed the first in situ analysis of human intestinal DCs and revealed
that in vivo populations in tissues differ from the widely used monocyte-derived DCs
generated in vitro
25
.
Therefore, it is important for a better understanding of the
pathophysiology of CD to characterise DC populations in colon and draining lymph nodes
in situ. On the basis of the in situ analysis of DC subpopulations, we can determine which
populations are of interest for future molecular characterisation. These DC populations may
be potential targets for future therapy.
DC marker Synonym
CD1a
BDCA1
BDCA2
BDCA3
CD1c
CD303
CD141
Cellular expression
Known or proposed function
Thymocytes, DCs (including
Recognition of non-protein lipid antigen, ligand for
Langerhans cells)
some gamma-delta T cells
Thymocytes, subsets of B cells,
Recognition of non-protein lipid antigen, ligand for
myeloid DCs
some gamma-delta T cells
Plasmacytoid DCs
Internalisation of antigen for presentation to T cells
Myeloid DCs
Activation of protein C
References
21
21
8, 24
22
Thrombomodulin
BDCA4
CD304
Neuropilin-1
CD83
CD209
s-100
DC-SIGN
Plasmacytoid DCs, endothelial cells
Neuronal receptor, co-receptor for vascular endothelial
growth factor A
Mature DCs
Co-stimulatory molecule
DCs, alveolar and decidual
Extravasation (ICAM-2), recognition of PAMPs,
macrophages
involved in T cell activation (ICAM-3)
Several nerve cell types, melanocytes,
Calcium-binding protein
Langerhans cells, DCs
Table 2. Cellular expression and known or proposed function of DCs present in tissues.
- 92 -
9
18
11, 20
7, 16, 23
DC populations in colon and MLNs of patients with CD
Materials and methods
Patients and tissue samples
Colon and MLNs were obtained with informed consent from patients with CD and nonIBD-related disorders (diverticulitis, polyposis coli, or colon carcinoma) by surgical
resection. Non-diseased colonic mucosa samples were obtained from patients with colon
cancer taken at least 7 cm from the tumour. MLNs that were devoid of cancer metastasis
were also obtained from these patients (CD; n=7) and non-IBD-related disorders (n=3).
Age range of the CD patients (n=9) was 26–41 years (mean age: 36 years), whereas the age
range of patients with non-IBD-related disorders (n=11) was 34–84 years (mean age: 57
years). Prior to the resection procedure, six of the nine CD patients were treated with
corticosteroids. After resection, colonic mucosa and MLNs were immediately snap frozen
in liquid nitrogen and stored at −80°C until cryostat sectioning. Alternatively, samples were
fixed in 4% buffered formaldehyde, dehydrated, and embedded in paraffin.
Immunohistochemistry
Frozen sections
BDCA-1-4 staining
Serial cryostat sections were cut on a Cryo-Star HM560 (Microm; Walldorf, Germany) and
transferred to aminopropyltriethoxysilane (APES)-coated glass slides (StarFrost; Knittel,
Klinipath, Duiven, The Netherlands), dried, and stored at −80C until further processing.
Tissue sections were defrosted at room temperature, dried, and fixed in ice-cold acetone for
10 minutes at room temperature. After fixation, tissue sections were rinsed with PBS (pH
7.8), placed in a semi-automatic stainer (Sequenza; Shandon, Breda, The Netherlands), and
incubated with normal goat serum (CLB; Amsterdam, The Netherlands) to block nonspecific staining for 10 minutes. Sections were then incubated with primary antibody for 60
minutes at room temperature. Mouse anti-human monoclonal antibodies (Miltenyi Biotec;
Bergisch Gladbach, Germany) directed against BDCA1 (AD5-8E7), BDCA2 (AC144),
BDCA3 (AD5-14H12), and BDCA4 (AD5-17F6) were used. All primary antibodies were
- 93 -
Chapter 4
diluted in PBS containing 1% blocking reagent (10,961,760; Roche Diagnostics GmbH,
Mannheim, Germany) to block endogenous avidin and biotin activity. Following incubation
with primary antibody, sections were rinsed with PBS for 5 minutes and incubated with
biotinylated goat anti-mouse antiserum (Biogenics; Klinipath) for 30 minutes at room
temperature. Next, sections were rinsed with PBS and incubated with streptavidin alkaline
phosphatase (ss-AP; Biogenics, Klinipath) for 30 minutes at room temperature. Sections
were then rinsed with PBS containing Tris buffer (0.2 mol/liter, pH 8.5) and incubated with
New Fuchsin (Chroma; Kongen, Germany) substrate (containing levamisole to block
endogenous AP enzyme activity) for 20 minutes at room temperature. Sections were
counterstained with Gill's haematoxylin, rinsed with distilled water, and mounted in Vecta
Mount (Vector Laboratories; Burlingame, CA). Control staining was performed by
substituting primary antibody with an isotypic control monoclonal antibody.
DC-SIGN and CD83 staining
Frozen sections were stained using a standard alkaline phosphatase protocol as described
previously
25
. In short, sections were incubated in 5% (v/v) normal goat serum (CLB) to
block non-specific staining for 10 minutes. Sections were then stained with either antiCD83 as primary MAb (Beckman Coulter; Mijdrecht, The Netherlands) or with FITClabelled anti-DC-SIGN (obtained from Dr. Y. van Kooyk; Free University Medical Centre,
Amsterdam, The Netherlands). Following incubation with primary antibody, sections were
extensively rinsed in PBS and incubated with biotinylated goat anti-mouse IgG (Dako;
Glostrup, Denmark). Next, FITC-labelled MAb sections were incubated with rabbit antiFITC antibody (Dako) and goat anti-rabbit antibody (Dako). Subsequently, sections were
incubated with streptavidin/biotin-conjugated alkaline phosphatase complex (ABC
protocol; Dako). Colour was developed using as substrate naphthol AS-MX phosphate (0.3
mg/ml) plus New Fuchsin (0.1 mg/ml; Chroma) in 0.2 M Tris–HCl buffer, pH 8.0 (ABC
protocol), and sections were counterstained with haematoxylin.
- 94 -
DC populations in colon and MLNs of patients with CD
Paraffin sections
Paraffin sections were stretched and dried at 37C overnight, deparaffinised in xylene, and
rehydrated in alcohol series. To block endogenous peroxidase activity, sections were treated
with 3% H2O2 in methanol for 20 minutes. Sections were subjected to heat-induced epitope
retrieval for 10 min at 95C. To block non-specific binding sites, sections were incubated
with normal goat serum (5% in PBS) for 10 minutes. Sections were then incubated with
primary antibodies CD1a (1:20; Dako) and s-100 (1:4000; Dako) diluted in PBS containing
5% BSA for 1 hour. After incubation with secondary antibodies (Dako), peroxidase activity
was demonstrated by using 3,3-diaminobenzidine tetrachloride (Sigma; St Louis, MO).
Finally, sections were counterstained with haematoxylin, dehydrated, and mounted in
Pertex (Sigma–Aldrich; Steinheim, Germany).
All sections were examined in a double-blind manner by a pathologist.
- 95 -
Chapter 4
Results
Myeloid DCs
Distribution patterns of myeloid DCs in colonic mucosa and MLNs of non-CD and CD
patients using the DC markers BDCA1, BDCA3, s-100, DC-SIGN, CD83, and CD1a are
summarised in Table 3.
Presence in colon
DC Marker
Localisation in colon in non-CD and CD patients
CD1a
absent
-
-
BDCA1
very few cells scattered throughout LP, mantle zone of follicles
+/-
+/-
BDCA2
few cells in lymph follicles
+/-
+/-
BDCA3
scattered throughout the LP and around blood vessels
++
++
BDCA4
high expression around blood vessels in LP
++
++
lymph follicles, scattered throughout LP and submucosa
+/-
++
CD83
1
1
NON-IBD
IBD
scattered throughout mucosa
+
+++
s-100
nerve vessels, DCs in lymph follicles, but not in the B cell follicles
+
++
DC Marker
Localisation in MLN in non-CD and CD patients
CD1a
absent
CD209
Presence in MLN
BDCA1
mantle zone of B cell follicles, very few cells scattered in medullary sinuses
BDCA2
few cells around HEVs
BDCA3
very high expression as a cell layer around (sub)capsular and medullary sinuses,
blood vessels, and B cell follicles
NON-IBD
IBD
-
-
+
+
+/-
+/-
+++
+++
high expression around high endothelial venules
++
++
paracortical areas
++
++
CD2091
high expression in periphery of the medullary cords
+++
+++
s-100
scattered throughout (para)cortical T cell areas, subcapsular (only CD patients)
+
++
BDCA4
CD83
1
Table 3. Localisation in colon and MLN of DC subtypes. 1 Earlier published by te Velde et al. 2003.
- 96 -
DC populations in colon and MLNs of patients with CD
BDCA1 and BDCA3
In colonic tissue, expression of BDCA1+ cells was mainly observed in association with
lymph follicles. Only a few cells were scattered throughout the lamina propria (LP) (see
figure 1a). In MLN, expression of BDCA1 was restricted to the mantle zone, and some
subsets of cells were scattered throughout medullary sinuses (see figure 1b). BDCA3
expression was found in single cells in the LP of both non-CD and CD patients (see figure
1c). Moreover, BDCA3 was expressed around blood vessels in the LP, submusosa, and
muscle layers of the colon. In MLN, BDCA3 was expressed around (sub)capsular and
medullary sinuses, blood vessels, and lymph follicles accentuating the frontier between Tand B-cell areas (see figure 1d).
s-100
In MLNs, s-100+ cells were scattered throughout the cortical and paracortical T-cell areas,
whereas cells located in B-cell areas did not express s-100 (see figure 2). S-100+ cells were
also found in paracortical sinuses in MLNs of CD patients, whereas the paracortical sinuses
of non-CD MLNs were completely devoid of s-100+ cells. The number of s-100+ cells was
increased in MLNs of CD patients (see table 3). Lymph follicles in colonic tissue
demonstrated a slight increase of s-100+ DCs in CD patients as compared with non-CD
patients. S-100+ DCs were also absent in the B-cell areas of the follicles.
DC-SIGN+ and CD83+ cells
It has been previously demonstrated that DC-SIGN+ cells are scattered throughout the
mucosa, and that the CD83+ population is present in aggregated lymphoid nodules and as
single cells in the LP
25
. In addition, we demonstrate in this present study that, in MLN,
expression of DC-SIGN is present in the periphery of medullary cords, which is populated
by macrophages and plasma cells. CD83+ cells were found in paracortical zones populated
by T cells (see figure 3). There were no differences between CD and non-CD patients.
- 97 -
Chapter 4
CD1a cells
Both colonic tissues and MLNs of non-CD and CD patients were completely negative for
CD1a expression (data not shown).
Plasmacytoid DCs
Plasmacytoid DCs are characterised by the expression of BDCA2 and BDCA4. BDCA2
expression of a few cells was found in lymph follicles in colon, whereas BDCA2+ cells
were absent in mucosa of both non-CD and CD patients (see figure 4). In contrast,
expression of BDCA4 was strong in endothelial cells in LP of both non-CD and CD
patients. In MLN, expression of both BDCA2 and BDCA4 was closely associated with
high endothelial venules (HEVs).
- 98 -
DC populations in colon and MLNs of patients with CD
Figure 1. Photographs of the immunohistochemical determination of the expression of the myeloid DC markers
BDCA1 (a,b) and BDCA3 (c,d) in colonic tissue (a,c) and MLNs (b,d) from CD patients. Bar = 100 m. The
inserts in b and d indicate the localisation of the staining in MLN.
Figure 2. Photographs of the immunohistochemical localisation of s-100 expression in colon (a,c) and MLN
(b,d) of non-CD (a,b) and CD patients (c,d). Bar = 100 m.
- 99 -
Chapter 4
Figure 3. Photographs of the immunohistochemical localisation of DC-SIGN (a) and CD83 (b) expression in
the MLN of CD patients. Bar = 100 m. The inserts indicate the localisation of staining in MLN.
Figure 4. Photographs of the immunohistochemical localisation of expression of the plasmacytoid DC markers
BDCA2 (a,b) and BDCA4 (c,d) in colonic tissue (a,c) and MLNs (b,d) of CD patients. Bar = 100 m. The insert
in a indicates the presence of BDCA2+ cells in follicles in the wall of the colon. Bar = 200m. The inserts in b
and d indicate the localisation of staining in MLN.
- 100 -
DC populations in colon and MLNs of patients with CD
Discussion
During an immune response, DCs traffic from peripheral tissues into draining lymph nodes
through lymphatic vessels. However, it is not yet known which DC populations are present
in the colon wall and which DC populations migrate from colonic tissue into the MLNs. In
humans, these DC populations may play an important role in the pathogenesis of CD.
Therefore, we investigated subtypes and their localisation of DCs in colon and MLN. We
demonstrate here that three different subpopulations of myeloid DCs populate the mucosa
of the colon and MLN of non-CD and CD patients. These populations consist of the
following: (1) immature DCs or macrophages that express DC-SIGN, (2) mature DCs that
express s-100 or CD83, and (3) mature DCs that express BDCA3. BDCA1 and CD1a
expressing DCs were virtually absent in colon as well as MLNs. Immature DCs and
macrophages are mainly localised at antigen-capturing sites such as the mucosa and
medullary cords, whereas mature DCs are present where antigen is presented, including the
T-cell areas in colonic lymph follicles and MLNs.
In general, lymph nodes contain three types of DCs, i.e., interdigitating DCs
(IDCs), follicular DCs, and plasmacytoid DCs. IDCs are like Langerhans cells
characterised by the expression of s-100, which is a member of the family of calciumbinding proteins
7,16,26
. Based on the localisation of markers, we demonstrated that s-100+
and also CD83+ cells present in the MLN are most likely IDCs, which are mature DCs from
myeloid origin and capable of presenting antigens to naive T cells in the paracortical areas.
Moreover, in colonic tissue, s-100 as well as CD83 demonstrated a similar expression
pattern in lymph aggregates, suggesting that they are involved in similar functional
processes. In contrast, s-100 expression in cervical lymph nodes was absent in lymph
follicles
23,27
. Furthermore, only subcapsular sinuses of CD patients and not of non-CD
patients harbour s-100+ DCs, indicating that in CD patients there is an increased influx of
mature DCs of MLNs via subcapsular sinuses into the T-cell areas. An increased number of
s-100+ DCs was also found in extrafollicular T-cell areas in hyperplastic palatine tonsils of
patients with tonsillitis
arthritis
29
28
, in several autoimmune diseases such as juvenile rheumatoid
and in primary biliary cirrhosis
30
confirming a role for s-100+ cells in
- 101 -
Chapter 4
inflammatory processes. Moreover, s-100 is expressed by nerve cells and cancer cells and is
used as a marker in cases of melanoma, nerve tissue cancer, and several carcinomas 7,26,31-35.
BDCA3+ cells are likely to be IDCs, but because of their specific localisation at
the border of lymph follicles and sinuses they seem to belong to a myeloid cell type other
than s-100+ and CD83+ DCs. These cells may be capable of interacting with B and T cells
to induce humeral responses. However, BDCA3+ cells may be more involved in antigencapturing processes because they are found in LP.
Follicular DCs are characterised by none of the described markers. Expression of
BDCA1 in the mantle zone of B-cell follicles may be indicative of follicular DCs; however,
expression is not restricted to DCs. B-cell subsets in the germinal centre and mantle zone of
lymph nodes, marginal zone B cells in the spleen, and a subpopulation of B cells in the
peripheral blood also express BDCA1
36-38
, which belongs to a family of lipid antigen-
39
presenting molecules . The observation that BDCA1+ cells were smaller and without the
characteristic morphology of DCs suggests that expression of BDCA1 in MLN is due to the
presence of B cells rather than DCs.
DC-SIGN is a C-type lectin involved in sampling antigens by recognition of
specific carbohydrate structures of autoantigens and foreign antigens and is therefore
mainly expressed by immature DCs and macrophages 11. This is consistent with our earlier
findings that DC-SIGN+ cells are scattered throughout colonic mucosa where these cells
can encounter antigens. DC-SIGN+ cells were present in medullary cords of MLNs, but in
contrast to cervical lymph nodes no expression was detected in the outer zone of the
paracortex 27.
Although CD1a is mainly used as a marker for Langerhans cells in the skin,
expression of this marker is not restricted to this cell type because subsets of interstitial and
monocyte-derived DCs also express CD1a
40-42
. However, CD1a was not detectable in
MLNs and colonic tissue of both non-CD and CD patients. Moreover, subsets of DCs in the
superficial lymph nodes were CD1a positive, whereas CD1a expression is absent in deeply
- 102 -
DC populations in colon and MLNs of patients with CD
located lymph nodes including mesenteric, mediastinal, and para-aortic lymph nodes
23
.
43
Additionally, expression of CD1a does not occur in inguinal and liver lymph nodes .
Expression of plasmacytoid DC marker BDCA2 has been observed in close
association with HEVs, indicating that tissue plasmacytoid DC precursors are bloodderived cells that enter MLN in the HEVs. In agreement with these results, Yoneyama et al.
demonstrated that myeloid DCs migrate via lymph vessels into draining lymph nodes,
whereas plasmacytoid DCs enter lymph nodes via the HEVs 24. BDCA2+ DCs seem to be
mainly involved in antigen-capturing processes, and BDCA2 is a C-type lectin
transmembrane glycoprotein that internalises antigens to present to T cells 8. BDCA2
expression was absent in colonic mucosa of both non-CD and CD patients, which coincides
with the results of Middel et al. 44. BDCA4 is a neuronal receptor of the class 3 semaphorin
family and is also expressed by endothelial cells to function as a coreceptor for vascular
endothelial growth factor A 9. This suggests that BDCA4 expression at HEVs and in the LP
is related to endothelial cells instead of DCs.
In the current study we have shown that different DC markers give variable
staining patterns so that there is no marker for DCs. These different ‘DC markers’ are
present in different sites in the colon and MLN in CD patients, indicating that DCs are a
heterogeneous group of antigen-presenting cells harbouring different functions. Our data
indicate that colonic tissue and MLNs harbour at least three different myeloid DC
populations. One population consists of immature DCs, which express DC-SIGN and is
mainly located at antigen-capturing sites in the mucosa and medullary cords. A second
population of DCs expresses BDCA3 and, similar to DC-SIGN+ DCs, is also present as
single cells in the mucosa, suggesting that BDCA3+ DCs may be involved in antigencapturing processes. In the MLN, their location around the lymph follicles suggests a role
in the activation of B and T cells. The third DC population consists of mature DCs that
express s-100 and CD83. Their location in the T-cell areas in both the colonic lymph
follicles as well as the MLN suggests that they have the capacity to present antigen to T
cells. DCs from plasmacytoid origin were hardly present in the colon and MLN. Although
- 103 -
Chapter 4
BDCA4 expression was found around the blood vessels, its expression is probably on
endothelial cells instead of DCs.
Future studies will involve the isolation of the described different DC populations
for further investigation to utilise favourable results for the development of new therapeutic
strategies in the treatment of CD.
Acknowledgements
We thank Esther de Groot and Trees Dellemijn for performing immunohistochemical
staining and Kate Cameron for carefully reading the manuscript.
- 104 -
DC populations in colon and MLNs of patients with CD
References
1 Iwasaki,A. (2007) Mucosal dendritic cells. Annu. Rev. Immunol. 25, 381-418
2 Lee,H.K. and Iwasaki,A. (2007) Innate control of adaptive immunity: dendritic cells and beyond. Semin.
Immunol. 19, 48-55
3 Hampe,J. et al. (2002) Association of NOD2 (CARD 15) genotype with clinical course of Crohn's disease: a
cohort study. Lancet 359, 1661-1665
4 Hugot,J.P. et al. (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease.
Nature 411, 599-603
5 Ogura,Y. et al. (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature
411, 603-606
6 Netea,M.G. et al. (2004) NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for
Crohn's disease. Eur. J. Immunol. 34, 2052-2059
7 Cochran,A.J. et al. (1993) S-100 protein remains a practical marker for melanocytic and other tumours.
Melanoma Res. 3, 325-330
8 Dzionek,A. et al. (2001) BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates
antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 194, 1823-1834
9 Dzionek,A. et al. (2002) Plasmacytoid dendritic cells: from specific surface markers to specific cellular
functions. Hum. Immunol. 63, 1133-1148
10 Fithian,E. et al. (1981) Reactivity of Langerhans cells with hybridoma antibody. Proc. Natl. Acad. Sci. U. S. A
78, 2541-2544
11 Geijtenbeek,T.B. et al. (2000) Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that
supports primary immune responses. Cell 100, 575-585
12 Grouard,G. et al. (1997) The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3
and CD40-ligand. J Exp. Med. 185, 1101-1111
13 Liu,Y.J. et al. (2001) Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol 2, 585-589
14 MacDonald,K.P. et al. (2002) Characterization of human blood dendritic cell subsets. Blood 100, 4512-4520
15 Rissoan,M.C. et al. (1999) Reciprocal control of T helper cell and dendritic cell differentiation. Science 283,
1183-1186
16 Takahashi,K. et al. (1984) Immunohistochemical localization and distribution of S-100 proteins in the human
lymphoreticular system. Am. J. Pathol. 116, 497-503
17 Valladeau,J. et al. (1999) The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of
Langerhans cells, and is rapidly internalized from the cell surface. Eur. J. Immunol. 29, 2695-2704
18 Zhou,L.J. and Tedder,T.F. (1995) Human blood dendritic cells selectively express CD83, a member of the
immunoglobulin superfamily. J. Immunol. 154, 3821-3835
- 105 -
Chapter 4
19 Baumgart,D.C. et al. (2005) Patients with active inflammatory bowel disease lack immature peripheral blood
plasmacytoid and myeloid dendritic cells. Gut 54, 228-236
20 Cambi,A. et al. (2005) How C-type lectins detect pathogens. Cell Microbiol. 7, 481-488
21 Jullien,D. et al. (1997) CD1 presentation of microbial nonpeptide antigens to T cells. J. Clin. Invest 99, 20712074
22 Sadler,J.E. (1997) Thrombomodulin structure and function. Thromb. Haemost. 78, 392-395
23 Takahashi,K. et al. (2001) Morphological interactions of interdigitating dendritic cells with B and T cells in
human mesenteric lymph nodes. Am. J. Pathol. 159, 131-138
24 Yoneyama,H. et al. (2004) Evidence for recruitment of plasmacytoid dendritic cell precursors to inflamed
lymph nodes through high endothelial venules. Int. Immunol. 16, 915-928
25 te Velde,A.A. et al. (2003) Increased expression of DC-SIGN+IL-12+IL-18+ and CD83+IL-12-IL-18dendritic cell populations in the colonic mucosa of patients with Crohn's disease. Eur. J. Immunol. 33, 143-151
26 Takahashi,K. et al. (1984) Immunohistochemical study on the distribution of alpha and beta subunits of S-100
protein in human neoplasm and normal tissues. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 45, 385-396
27 Engering,A. et al. (2004) Dynamic populations of dendritic cell-specific ICAM-3 grabbing nonintegrin-positive
immature dendritic cells and liver/lymph node-specific ICAM-3 grabbing nonintegrin-positive endothelial cells in
the outer zones of the paracortex of human lymph nodes. Am. J. Pathol. 164, 1587-1595
28 Papadas,T. et al. (2001) S-100 protein-positive dendritic cells and CD34-positive dendritic interstitial cells in
palatine tonsils. Eur. Arch. Otorhinolaryngol. 258, 243-245
29 Frosch,M. et al. (2003) Expression of myeloid-related proteins 8 and 14 in systemic-onset juvenile rheumatoid
arthritis. Arthritis Rheum. 48, 2622-2626
30 Demetris,A.J. et al. (1989) S100 protein positive dendritic cells in primary biliary cirrhosis and other chronic
inflammatory liver diseases. Relevance to pathogenesis? Am. J. Pathol. 134, 741-747
31 Daimaru,Y. et al. (1985) Malignant peripheral nerve-sheath tumors (malignant schwannomas). An
immunohistochemical study of 29 cases. Am. J. Surg. Pathol. 9, 434-444
32 Drier,J.K. et al. (1987) S100 protein immunoreactivity in poorly differentiated
Immunohistochemical comparison with malignant melanoma. Arch. Pathol. Lab Med. 111, 447-452
carcinomas.
33 Gillett,C.E. et al. (1990) S100 protein in human mammary tissue--immunoreactivity in breast carcinoma,
including Paget's disease of the nipple, and value as a marker of myoepithelial cells. J. Pathol. 160, 19-24
34 Monda,L. and Wick,M.R. (1985) S-100 protein immunostaining in the differential diagnosis of
chondroblastoma. Hum. Pathol. 16, 287-293
35 Wick,M.R. et al. (1988) Recognition of malignant melanoma by monoclonal antibody HMB-45. An
immunohistochemical study of 200 paraffin-embedded cutaneous tumors. J. Cutan. Pathol. 15, 201-207
36 Plebani,A. et al. (1993) B and T lymphocyte subsets in fetal and cord blood: age-related modulation of CD1c
expression. Biol. Neonate 63, 1-7
37 Small,T.N. et al. (1987) M241 (CD1) expression on B lymphocytes. J. Immunol. 138, 2864-2868
- 106 -
DC populations in colon and MLNs of patients with CD
38 Smith,M.E. et al. (1988) CD1c antigens are present in normal and neoplastic B-cells. J. Pathol. 156, 169-177
39 Brigl,M. and Brenner,M.B. (2004) CD1: antigen presentation and T cell function. Annu. Rev. Immunol. 22,
817-890
40 Tazi,A. et al. (1993) Evidence that granulocyte macrophage-colony-stimulating factor regulates the distribution
and differentiated state of dendritic cells/Langerhans cells in human lung and lung cancers. J. Clin. Invest 91, 566576
41 Yoshida,A. et al. (1997) Increased number of IgE positive Langerhans cells in the conjunctiva of patients with
atopic dermatitis. Br. J. Ophthalmol. 81, 402-406
42 Zhou,L.J. and Tedder,T.F. (1996) CD14+ blood monocytes can differentiate into functionally mature CD83+
dendritic cells. Proc. Natl. Acad. Sci. U. S. A 93, 2588-2592
43 Tanis,W. et al. (2004) Human hepatic lymph nodes contain normal numbers of mature myeloid dendritic cells
but few plasmacytoid dendritic cells. Clin. Immunol. 110, 81-88
44 Middel,P. et al. (2006) Increased number of mature dendritic cells in Crohn's disease: evidence for a chemokine
mediated retention mechanism. Gut 55, 220-227
- 107 -
Chapter 4
- 108 -
Chapter
5.
Single nucleotide polymorphisms in Ctype lectin genes, clustered in the IBD2
and IBD6 susceptibility loci, may play
a role in the pathogenesis of
inflammatory bowel diseases
Simone C. Wolfkamp1,2, Marleen I. Verstege1,2, Sander Meisner2, Pieter C.
Stokkers1, Anje A. te Velde2
1. Department of Gastroenterology and Hepatology, Academic Medical Centre,
Amsterdam
2. Tytgat Institute for Liver and Intestinal Diseases, Academic Medical Centre,
Amsterdam
Chapter 5
Abstract
The balance between microbes and host defence mechanisms at the mucosal frontier
plays an important, yet unclarified role in the pathogenesis of inflammatory bowel
disease (IBD). The importance of micro-organisms in IBD is supported by the
association of IBD with mutations in pathogen recognition receptors (PRRs) as
NOD2 and TLR4. We wanted to investigate whether polymorphisms in another type
of PRRs, the so-called C-type lectin receptors (CLRs) are associated with IBD.
Growing insight in the pathogenetic role of NOD2 mutations in Crohn´s disease
(CD) and the fact that the majority of CLR-encoding genes are located in IBD
susceptibility loci provide strong arguments for further exploration of the role of
CLRs in IBD. In this study, we selected four single nucleotide polymorphisms
(SNPs) in different CLRs to see whether there could be a role for these CLRs in
IBD. Functional SNPs in the candidate CLRs DC-SIGN, LLT1, DCIR and MGL
were investigated. Genotyping of all SNP’s was performed at the AMC. In this
study a total of 1348 patients were included of which 535 CD patients, 371
ulcerative colitis (UC) patients and 442 healthy controls (HC). No association was
found between our IBD cohort and the candidate SNPs for DC-SIGN (CD/HC:
p=0.25 and UC/HC: p=0.36), DCIR (CD/HC: p=0.22 and UC/HC: p=0.41) and
MGL (CD/HC: p=0.37 and UC/HC: p=0.25). However, polymorphisms in LLT1
were associated with our CD population (p<0.034). Our UC cohort was not
associated with the variation in LLT1 (p=0.33). LLT1 is a ligand for recently
discovered CD161. CD161 is a new surface marker for human IL-17 producing
Th17 cells. The Th17 phenotype has been linked to CD by the fact that IL-22, IL-17
and IL-23 receptor levels are increased in CD. The signal transduction pathways
involving LLT1 and CD161 are not completely clarified and currently under
investigation in our laboratory.
- 110 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
Introduction
Inflammatory Bowel Disease (IBD) is comprised of two major disorders, knowing
Crohn’s Disease (CD) and Ulcerative Colitis (UC). Both diseases are characterised
by chronic, relapsing intestinal inflammation causing diarrhoea, abdominal pain and
malnutrition. CD can affect any part of the gastrointestinal tract, although the most
common areas are the terminal ileum and colon, whereas UC is limited to the colon.
Within the last era it has become clear that next to immunological and
environmental factors, genetic factors play an important role in the pathogenesis of
IBD. It is suggested that there is an inadequate immune response in a genetic
susceptible host. Antigen presenting cells (APCs) highly express pattern recognition
receptors (PRRs) to detect highly conserved structures of microbes and altered self
proteins. Dependent on which PRRs are activated by specific antigens, APCs will
direct naïve T cells to differentiate into Th1, Th2, Th17 of regulatory T cells. It has
been shown earlier that mutations in the PRRs NOD2 (CARD15), NALP3 (CIAS1,
NLRP3) and Toll-like receptor-4 are associated with the development of CD 1-4.
One of the major classes of the PRRs are the C-type lectins. C-type lectin
like receptors (CLRs) are expressed on dendritic cells (DCs) and macrophages,
reflecting the position of these cell types as sensors at the interface of the immune
system and environment. In 1860 the first animal lectins were already described as
being a substance of snake venom, but it was Drickamer in 1988 who suggested a
categorisation of the lectins into structural categories 5,6. Today, the classification of
lectins is based on the primary protein sequence of the carbohydrate recognition
domain (CRD), the protein domain involved in carbohydrate binding 6,7 and the Ca2+
mediated carbohydrate recognition, typical for CLRs. CLRs are folded according the
specific C-type lectin-like fold, consisting of two anti-parallel β strands and two α
helices
7,8
. They can be divided into two categories based on Ca2+ ion coordination
and the orientation of their N-terminus. Although carbohydrate recognition is
primarily determined by the amino acid sequence of the CRD fold, it is strongly
influenced by the oligomerisation of the receptor. Oligomerisation not only
- 111 -
Chapter 5
strengthens the binding to a certain structure, it also limits the binding to ligands
with a complementary carbohydrate density and spacing 9. Most CLRs are present as
transmembrane proteins on APCs, mostly immature DCs, although they can also
appear soluble in the cell 8. Antigens can be taken up through CLRs, processed and
presented to major histocompatibility complex (MHC) classes I and II, thereby
inducing CD4 and CD8 T-cell responses. CLRs also play a role in signalling
pathways upon glycan interaction and can thereby mediate a pro- or antiinflammatory response 7. These responses are also influenced by the presence of a
sequence motif, found in the intracellular domain of various receptors. The motifs,
termed immunoreceptor tyrosine based activation (or inhibitory) motifs, (ITAMs or
ITIMs), provide the basis for two opposed signalling modules that control the
balance of the immune silencing and activation
10
. CLRs have been shown to be
involved in several aspects of the immune response. Growing insight in the
pathogenetic role of mutations in PRRs as NOD2 in CD and the fact that the
majority of the CLR encoding genes are located in IBD susceptibility loci 2 and 6
provide strong arguments for further exploration of the role of CLRs in IBD. We
selected four CLRs in this study to see whether there could be a role for these CLR’s
in IBD. All four selected CLRs, knowing DC-SIGN (DC-specific intercellular
adhesion molecule 3 (ICAM-3) grabbing nonintegrin), MGL (Macrophage
Galactose-type Lectin, CLEC10A, CD301), DCIR (Dendritic Cell Immunoreceptor,
CLEC4A) and LLT1 (Lectin-like transcript 1, CLEC2D) were chosen based on their
known function, their presence on a susceptibility locus and their association with
other chronic inflammatory or autoimmune diseases.
- 112 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
Materials and Methods
Genotyping
In this study we genotyped our cohort for four SNP’s in different CLRs (see table 1).
DNA was isolated from venous blood, which was collected over the years from
patients during periodic visits. Genotyping for the SNP’s was performed by
polymerase chain reaction restriction fragment length polymorphisms (PCR-RFLP).
Designed primers, thermal cycling and restriction enzymes (all from New England
BioLabs, Ipswich, MA) are listed in table 1. Restriction fragments were separated
and visualised using 3% agarose gel containing ethydium bromide.
CLEC4M
CLEC10a
CLEC4a
CLEC2d
C-type lectin
DC-SIGN
MGL
DCIR
LLT1
rs number
rs735239
rs90951
rs2024301
rs3764022
Forward:
3’ccaccagcaggtgaatga
ta 5’
Forward:
3’ggggtgtgaggggcctgtgtg
actgaggac 5’
Forward:
3’cacagtggattcccatgaatta
tgtctccccagaga 5’
Forward:
3’ggtagctttaatagaatgctctt
ttgaatgca 5’
Reverse:
3’ccacctgctccttcctgata
5’
Reverse:
3’ggacagcaggagatggcag
ggcccagacc 5’
Reverse:
3’cttcttcttcagcttccaaggag
aggactgcccct 5’
Reverse:
3’cagttacttacttggattttgttg
atttcattcct 5’
96°C 15 min
96°C 30 sec |
57°C 1 min }35x
72°C 1 min |
72°C 10 min
8°C ∞
95˚C 5 min
94°C 30 sec |
57°C 45 sec }33x
72°C 45 sec |
72°C 5 min
10°C ∞
95˚C 5 min
94°C 30 sec |
58°C 45 sec }33x
72°C 45 sec |
72°C 5 min
10°C ∞
95˚C 5 min
94°C 30 sec |
58°C 45 sec }33x
72°C 45 sec |
72°C 5 min
10°C ∞
Primers
Thermal
cycling
Restriction
enzymes
HpyCH4 IV
AvaI
MaeIII
Bsm AI
Product size
240 bp
275 bp
272 bp
328 bp
Restriction
digestion
patterns
GG:240 bp
AG: 240, 143, 96 bp
AA: 143, 96 bp
AA: 275 bp
AG: 275, 246, 29 bp
GG: 246, 29 bp
AA: 272 bp
AT: 272, 235, 37 bp
TT: 235, 37 bp
GG: 396, 32 bp
GC: 328, 396, 32 bp
CC: 396, 32 bp
Table 1. Designed primers, thermal cycling and restriction enzymes.
- 113 -
Chapter 5
Patients and controls for genotyping
The study population consisted of 1572 IBD patients. All patients were recruited
through the outpatient clinic at the department of Gastroenterology in the Academic
Medical Centre Amsterdam, Netherlands as part of an ongoing project to investigate
genetic factors that contribute to the aetiology of IBD. All phenotypic patient
information has been collected in an electronic patient file. The control group
consisted of in total 586 healthy volunteers, partially the partner of the patient. All
patients and controls gave informed consent and the study was approved by the
ethics review committees of each of the participating university hospitals. All DNA
samples and data in this study were handled anonymously.
Statistics
Confirmation to Hardy-Weinberg equilibrium was tested using 2 test. Genotypes
and allele frequencies of patients and HCs were compared by 2 test. Significance
level was set at p<0.05. All data were analysed using Graphpad prism 4 (Graphpad
Prism v. 4 for Windows, GraphPad Software, San Diego, California USA).
- 114 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
Results
DC-SIGN
In total 1354 samples were genotyped for DC-SIGN (SNP rs735239) of which were
768 IBD patients (386 CD patients vs. 382 UC patients) and 586 healthy controls
(HCs). When CD patients were compared to HCs no significant difference between
the genotype frequencies was found (p=0.25), with G-allele frequencies of 65.5%
for CD patients and 69.1% for HCs (p=0.10) (see table 2a). Moreover, analysis of
the genotype frequencies between UC patients and HCs showed no significant
difference (p=0.36), with G-allele frequencies of 66.1% for UC patients and 69.1%
for HCs (p=0.16) (see table 2b).
MGL
In total 1572 samples were genotyped for MGL (SNP rs90951) of which were 1017
IBD patients (560 CD patients vs. 457 UC patients) and 555 HCs. When CD patients
were compared to HCs no significant difference between the genotype frequencies
was found (p=0.37), with A-allele frequencies of 66.7% for CD patients and 63.9%
for HCs (p=0.16) (see table 2a). Besides, analysis of the genotypes frequencies
between the UC patients and the HCs showed no significant difference (p=0.25),
with A-allele frequencies of 66.7% for UC patients and 63.9% for HCs (p=0.16) (see
table 2b).
DCIR
In total 1349 samples were genotyped for DCIR (SNP rs2024301) of which were
830 IBD patients (531 CD patients vs. 299 UC patients) and 519 HCs. When CD
patients were compared to HCs no significant difference between the genotype
frequencies was found (p=0.22), with A-allele frequencies of 66.9% for CD patients
and 63.6% for HCs (p=0.12) (see table 2a). Moreover, analysis of the genotypes
frequencies between the UC patients and the HCs showed no significant difference
- 115 -
Chapter 5
(p=0.41), with A-allele frequencies of 66.7% for UC patients and 63.6% for HCs
(p=0.20) (see table 2b).
LLT1
In total 1511 samples were genotyped for LLT1 (SNP rs3764022) of which were
1025 IBD patients (621 CD patients vs. 404 UC patients) and 486 HCs. When CD
patients were compared to HCs a significant difference between the genotype
frequencies was found with a p-value of 0.03. However, comparison of the allele
frequencies (G-allele: 68.0% for CD and 65.6% for HCs) showed no significant
difference (p=0.23) (see table 2a). Analysis of the genotype frequencies between the
UC patients and the HCs showed no significant difference (p=0.33). When allele
frequencies were assessed, no significant difference (p=0.52) was found (G-allele:
67.1% for UC patients and 65.6% for HCs) (see table 2b).
- 116 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
CD
1
DC-SIGN
MGL
DCIR
LLT1
HC
Genotype n (%)
2
Allelefreq. n (%)
1
P –value
Genotype n (%)
2
Allefreq. n (%)
GG
166 (43,0)
G
506 (65,5)
GG
283 (48,3)
G
810 (69,1)
1
P = 0.25
GA
174 (45,1)
A
266 (34,5)
GA
244 (41,6)
A
362 (30,9)
2
P = 0.10
AA
46 (11,9)
AA
59 (10,1)
AA
256 (45,7)
A
747(66,7)
AA
237 (42,7)
A
709(63,9)
1
P=0.37
AG
235 (42,0)
G
373 (33,3)
AG
235 (42,3)
G
401 (36,1)
2
P = 0.16
GG
69 (12,3)
GG
83 (15,0)
AA
239 (45,0)
A
710 (66,9)
AA
218 (42,0)
A
660 (63,6)
1
P=0.22
AT
232 (43,7)
T
352 (33,1)
AT
224 (43,2)
T
378 (36,4)
2
P=0.12
TT
60 (11,3)
TT
77 (14,8)
GG
277(44,6)
G
845 (68,0)
GG
216 (44,4)
G
638 (65,6)
1
P=0.03*
GC
291 (46,9)
C
397 (32,0)
GC
206 (42,4)
C
334 (34,4)
2
P=0.23
CC
53 (8,5)
CC
64 (13,2)
Table 2a. Genotype frequencies and allele frequencies of CD patients compared to HCs. n =
number of patients, % = percentage, 1 p = genotype frequency p-value, 2 p = allele frequency pvalue. - 117 -
Chapter 5
UC
1
DC-SIGN
MGL
DCIR
LLT1
HC
Genotype n (%)
2
Allelefreq. n (%)
1
P –value
Genotype n (%)
2
Allefreq. n (%)
GG
167 (43,7)
G
505 (66,1)
GG
283 (48,3)
G
810 (69,1)
1
P = 0.36
GA
171 (44,8)
A
259 (33,9)
GA
244 (41,6)
A
362 (33,9)
2
P = 0.16
AA
44 (11,5)
AA
59 (10,1)
AA
205 (44,9)
A
610 (66,7)
AA
237 (42,7)
A
709 (63,9)
1
P= 0.25
AG
200 (43,8)
G
304 (33,3)
AG
235 (42,3)
G
401 (36,1)
2
P= 0.16
GG
52 (11,4)
GG
83 (15,0)
AA
135 (45,2)
A
399 (66,7)
AA
218 (42,0)
A
660 (63,6)
1
P = 0.41
AT
129 (43,1)
T
199 (33,3)
AT
224 (43,2)
T
378 (36,4)
2
P = 0.20
TT
35 (11,7)
TT
77 (14,8)
GG
179 (44,3)
G
542(67,1)
GG
216 (44,4)
G
638(65,6)
1
P= 0.33
GC
184 (45,5)
C
266(32,9)
GC
206 (42,4)
C
334(34,4)
2
P = 0.52
CC
41 (10,1)
CC
64 (13,2)
Table 2b. Genotype frequencies and allele frequencies of UC patients compared to HCs. n =
number of patients, % = percentage, 1 p = genotype frequency p-value, 2 p = allele frequency pvalue.
- 118 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
Discussion and Conclusion
In this study we hypothesised that mutations in CLRs as innate sensors of DCs are
associated with IBD. All chosen CLRs are located on IBD susceptibility loci and are
associated with other chronic inflammatory or autoimmune diseases. The most
studied CLR of the four is the Ca2+-dependent type II C-type lectin DC-SIGN,
encoded by a member of the CD209 gene family on chromosome 19p13
6,7
. This
receptor binds the natural ligands ICAM-2 on endothelial cells and ICAM-3 on T
cells resulting in close cell-cell contact, required for extravasation and antigen
presentation respectively 11-13. Moreover, it may represent a common thread in many
inflammatory diseases as IBD, as it has also been shown to facilitate direct infection
of DCs by a range of infectious agents, such as HIV-1, Dengue virus, Ebola virus,
human cytomegalovirus, Leishmania pifanoi amastigotes and Mycobacterium
tuberculosis 14. Studies using anti-DC-SIGN antibodies as antigen indicate that DCSIGN functions as a receptor that mediates antigen uptake for presentation to T cells
15
. Hardly any data demonstrate that pathogen internalisation through DC-SIGN
leads to enhanced pathogen-specific T cell responses. The general finding is that
several pathogens target CLRs on DCs to evade immune responses, such as HIV-1
or the secreted product ManLAM of M. tuberculosis that inhibits DC maturation and
enhances the production of IL-10
. Lactobacilli modulate DCs to stimulate the
16,17
development of regulatory T cells via DC-SIGN
18
. In addition, other bacteria
binding to DC-SIGN induce a skewing of the immune response to a Th1- or Th2like cytokine profile 19,20. The general thought is that antigen uptake via CLRs in the
absence of danger signals via Toll-like receptors does not lead to effective
immunity, but instead to the induction of tolerogenic T cells 21. It has been suggested
by Nunez et al., that another functional variant in the CD209 gene, rs4804803, could
be involved in the aetiology or pathology of UC in HLA-DR3-positive individuals.
These findings suggest a role potential role for DC-SIGN in the pathophysiology of
IBD. However, in this study we were unable to find associations between
polymorphisms in DC-SIGN and any of our IBD patient groups.
- 119 -
Chapter 5
Macrophage galactose-type lectin (MGL, CLEC10A, CD301), located on
chromosome 17p13, is a CLR expressed on monocyte-derived immature DCs and an
intermediate stage of macrophage differentiation
22,22-24
. It has been identified on
dermal, gut, thymus and lymph node APCs. MGL is not expressed by monocytes,
lymphocytes, plasmacytoid DC’s and resident Langerhans cells 24. Moreover, MGL
is upregulated during chronic inflammatory conditions such as rheumatoid arthritis
on APCs in the affected tissue, maybe linking MGL to IBD. MGL has a specific
CRD for terminal Ga1NAc moieties, parts of glycoproteins or glycolipids that
function as ligands for MGL
25
. As PRR, MGL can bind to CD45, thereby down-
regulating the T cell activation via APCs, which results in decreasing cytokine and
proliferative responses and T cell death
26
. Homeostatic control of T cells involves
tight regulation of effector T cells to prevent excessive activation that can cause
tissue damage and autoimmunity, also thereby linking MGL to IBD as an
autoimmune disease. Up until today there are no signalling pathways established for
MGL. However, van Vliet et al. suggested that MGL may play a role in negative
regulation of effector T-cells and negatively regulates DC maturation
26,27
. The fact
that MGL can be linked to both chronic inflammatory diseases as well as
autoimmune diseases resulted in this study to associate SNPs in MGL with IBD.
Unfortunately, we were unable to detect an association of polymorphisms in MGL
with our IBD patient cohort.
DCIR (CLED4A) located on chromosome 12p13, was originally presented
by Bates in 1999 as molecular homologue of asialoglycoprotein receptor (ASGPR)
and macrophage lectin
11,28
. It is a C-type lectin expressed on DC’s, granulocytes,
monocytes, macrophages and B-cells. DCIR has an ITIM motif, which has shown to
inhibit signal transduction when activated and it is present in the cytoplasmic tail of
the C-type lectin-like molecules. After stimulation with LPS or the proinflammatory cytokines TNF-and IL-1 DCIR was down-regulated during
inflammation, although stimulation with other cytokines as GM-CSF, IL-3, IL-4 and
IL-13 showed accumulation of a short form of DCIR mRNA, which encodes a
putative non-functional protein which may act as an antagonist to the full length
- 120 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
receptor
29,30
. The presence of the ITIM and the down-regulation of DCIR in pro-
inflammatory settings, suggest a regulatory role for this receptor
31
. Furthermore,
Fujikado et al. showed that DCIR also plays a role in controlling autoimmune
diseases 28. Aged DCIR-deficient mice developed joint abnormalities, had elevated
levels of auto-antibodies and showed higher levels of CD11c+ DCs and proportional
expansion of T cell populations in their lymph nodes. Young DCIR-deficient mice
were found to develop more severe disease than their wild type littermates,
suggesting a protective effect of DCIR
31,32
. Also, the DCIR receptor was found
abundantly in synovial fluid of patients with rheumatoid arthritis. This was the
opposite in the control group, suggesting a role for DCIR in the development of
autoimmune diseases 31. These last two findings suggest both an anti-inflammatory
as well as an autoimmune role for DCIR, thereby linking it to IBD. We were unable
to establish a role for variants of DCIR in our IBD patient cohort.
The last CLR genotyped for its SNP was LLT1, also known as CLEC2D.
This CLR is located on IBD locus 2 and is expressed on activated B cells and DCs.
Furthermore, LLT1 is expressed on peripheral blood mononuclear cells after
stimulation through several Toll-like receptors. We found an association between
LLT1 and our CD population (p<0.034). Since the homozygous mutant is less
common in the CD cohort, it is likely that this polymorphism protects against the
development of CD. Our UC cohort was not associated with the variation in LLT1
(p=0.33). LLT1 is a ligand for CD161 and as a complex it inhibits NK cell-mediated
cytotoxicity and cytokine production 33. CD161 is a new surface marker for human
IL-17 producing Th17 cells
34,35
. The Th17 phenotype has recently been linked to
CD by the fact that IL-22, IL-17 and IL-23 receptor levels are increased in CD 36.
Further studies will analyse the role of CD161 in our IBD patient cohort.
Although we were unable to associate three of the four candidate SNPs to
our IBD patient cohort, this does not mean that CLRs do not play a role in the
pathophysiology of IBD. This is illustrated by the recent finding of Thebault et al. 37
who demonstrate that CLEC-1 is associated with a higher production of IL-17.
- 121 -
Chapter 5
CLEC-1, expressed by myeloid cells and endothelial cells, is enhanced by regulatory
mediators and moderates Th17 differentiation and thereby involved in processes of
autoimmunity and thus IBD. Further studies and extensive SNP analysis in ongoing
Genome Wide Association Studies will clarify the role of CLRs in autoimmunity.
Acknowledgements
Research performed by S.W. was funded by the Dutch digestive disease foundation.
- 122 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
References
1 Hugot,J.P. et al. (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's
disease. Nature 411, 599-603
2 Ogura,Y. et al. (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease.
Nature 411, 603-606
3 Braat,H. et al. (2005) Consequence of functional Nod2 and Tlr4 mutations on gene transcription in
Crohn's disease patients. J. Mol. Med. 83, 601-609
4 Villani,A.C. et al. (2009) Common variants in the NLRP3 region contribute to Crohn's disease
susceptibility. Nat. Genet. 41, 71-76
5 Zelensky,A.N. and Gready,J.E. (2005) The C-type lectin-like domain superfamily. FEBS J. 272, 61796217
6 Drickamer,K. (1988) Two distinct classes of carbohydrate-recognition domains in animal lectins. J.
Biol. Chem. 263, 9557-9560
7 van Vliet,S.J. et al. (2008) Dendritic cells and C-type lectin receptors: coupling innate to adaptive
immune responses. Immunol. Cell Biol. 86, 580-587
8 Figdor,C.G. et al. (2002) C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev.
Immunol. 2, 77-84
9 Weis,W.I. et al. (1998) The C-type lectin superfamily in the immune system. Immunol. Rev. 163, 19-34
10 Billadeau,D.D. and Leibson,P.J. (2002) ITAMs versus ITIMs: striking a balance during cell
regulation. J. Clin. Invest 109, 161-168
11 Bates,E.E. et al. (1999) APCs express DCIR, a novel C-type lectin surface receptor containing an
immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 163, 1973-1983
12 Geijtenbeek,T.B. et al. (2000) DC-SIGN-ICAM-2 interaction mediates dendritic cell trafficking. Nat.
Immunol. 1, 353-357
13 Geijtenbeek,T.B. et al. (2000) Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3
receptor that supports primary immune responses. Cell 100, 575-585
14 Geijtenbeek,T.B. et al. (2004) Self- and nonself-recognition by C-type lectins on dendritic cells. Annu.
Rev. Immunol. 22, 33-54
15 Engering,A. et al. (2002) The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen
for presentation to T cells. J. Immunol. 168, 2118-2126
16 Arrighi,J.F. et al. (2004) DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1
transmission from dendritic cells to T cells. J. Exp. Med. 200, 1279-1288
17 Geijtenbeek,T.B. et al. (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. J.
Exp. Med. 197, 7-17
- 123 -
Chapter 5
18 Smits,H.H. et al. (2005) Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro
by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3grabbing nonintegrin. J. Allergy Clin. Immunol. 115, 1260-1267
19 Bergman,M.P. et al. (2004) Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance
through phase-variable interaction between lipopolysaccharide and DC-SIGN. J. Exp. Med. 200, 979-990
20 Steeghs,L. et al. (2006) Neisseria meningitidis expressing lgtB lipopolysaccharide targets DC-SIGN
and modulates dendritic cell function. Cell Microbiol. 8, 316-325
21 Bonifaz,L. et al. (2002) Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in
the steady state leads to antigen presentation on major histocompatibility complex class I products and
peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627-1638
22 Higashi,N. et al. (2002) Human macrophage lectin specific for galactose/N-acetylgalactosamine is a
marker for cells at an intermediate stage in their differentiation from monocytes into macrophages. Int.
Immunol. 14, 545-554
23 van Vliet,S.J. et al. (2008) Sweet preferences of MGL: carbohydrate specificity and function. Trends
Immunol. 29, 83-90
24 Sandra J.van Vliet (7 A.D.) Novel insights into MGL-glycan interactions in the immune system
25 van Vliet,S.J. et al. (2005) Carbohydrate profiling reveals a distinctive role for the C-type lectin MGL
in the recognition of helminth parasites and tumor antigens by dendritic cells. Int. Immunol. 17, 661-669
26 van Vliet,S.J. et al. (2006) Regulation of effector T cells by antigen-presenting cells via interaction of
the C-type lectin MGL with CD45. Nat. Immunol. 7, 1200-1208
27 van Vliet,S.J. et al. (2008) The C-type lectin macrophage galactose-type lectin impedes migration of
immature APCs. J. Immunol. 181, 3148-3155
28 Kanazawa,N. (2007) Dendritic cell immunoreceptors: C-type lectin receptors for pattern-recognition
and signaling on antigen-presenting cells. J. Dermatol. Sci. 45, 77-86
29 Richard,M. et al. (2002) The expression pattern of the ITIM-bearing lectin CLECSF6 in neutrophils
suggests a key role in the control of inflammation. J. Leukoc. Biol. 71, 871-880
30 Richard,M. et al. (2003) The ITIM-bearing CLECSF6 (DCIR) is down-modulated in neutrophils by
neutrophil activating agents. Biochem. Biophys. Res. Commun. 310, 767-773
31 Graham,L.M. and Brown,G.D. (2009) The Dectin-2 family of C-type lectins in immunity and
homeostasis. Cytokine
32 Fujikado,N. et al. (2008) Dcir deficiency causes development of autoimmune diseases in mice due to
excess expansion of dendritic cells. Nat. Med. 14, 176-180
33 Rosen,D.B. et al. (2008) Functional consequences of interactions between human NKR-P1A and its
ligand LLT1 expressed on activated dendritic cells and B cells. J. Immunol. 180, 6508-6517
34 Cosmi,L. et al. (2008) Human interleukin 17-producing cells originate from a CD161+CD4+ T cell
precursor. J. Exp. Med. 205, 1903-1916
- 124 -
SNPs in C-type lectin genes may play a role in the pathogenesis of IBD
35 Kleinschek,M.A. et al. (2009) Circulating and gut-resident human Th17 cells express CD161 and
promote intestinal inflammation. J. Exp. Med. 206, 525-534
36 Fina,D. et al. (2008) Regulation of gut inflammation and th17 cell response by interleukin-21.
Gastroenterology 134, 1038-1048
37 Thebault,P. et al. (2009) The C-type lectin-like receptor CLEC-1, expressed by myeloid cells and
endothelial cells, is up-regulated by immunoregulatory mediators and moderates T cell activation. J.
Immunol. 183, 3099-3108
- 125 -
Chapter 5
- 126 -
Part 3
Cholinergic modulation of
intestinal immune
responses
Chapter
6.
The enteric nervous system as
regulator of intestinal epithelial barrier
function in health and disease
Susanne A. Snoek1, Marleen I. Verstege1, Guy E. Boeckxstaens1,2, Rene M. van
den Wijngaard1, Wouter J. de Jonge1
1.Tytgat institute of liver and intestinal research, Academic Medical Centre,
Amsterdam
2. Department of Gastroenterology, Catholic University of Leuven, Leuven,
Belgium
Chapter 6
Abstract
The intestinal epithelia proliferate and differentiate along the crypt villus axis to
constitute a barrier cell layer separating some 1013 potentially harmful bacteria from
a sterile mucosal compartment. Strict regulatory mechanisms are required to
maintain a balance between appropriate uptake of luminal food components and
proteins, while constraining exposure of the mucosal compartment to luminal
antigens and microbes. The enteric nervous system is increasingly recognised as
such as regulatory housekeeper of epithelial barrier integrity, in addition to its
ascribed immunomodulatory potential. Inflammation affects both epithelial integrity
and barrier function and in turn, loss of barrier function perpetuates inflammatory
conditions. The observation that inflammatory conditions affect enteric neurons may
add to the dysregulated barrier function in chronic disease.
Here, we review recent advances in the understanding of the regulatory role
of the nervous system in the maintenance of barrier function in healthy state, or
during pathological conditions of for instance stress induced colitis, surgical trauma,
or inflammation. We will discuss clinical potential of the advances in understanding
of the role of the enteric nervous systems in this important phenomenon.
- 130 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Introduction and Scope
Homeostasis of the intestinal barrier is essential to give access of nutrients and
electrolytes into the interior milieu; while at the same time should control passage of
pathogens. Regulation of the intestinal barrier function involves the interplay of
several mechanisms, such as integrity of the epithelial layer, and a specialised
immune system for surveillance, regulated phagocytosis, killing and processing of
(commensal) bacteria. Disturbances in the homeostatic conditions of the intestinal
barrier are an important factor in the initiation and perpetuation of pathological
conditions including inflammatory bowel diseases (IBD); stress-induced disorders
such as irritable bowel syndrome (IBS) and food allergies; pathogen-induced
intestinal inflammation and septic morbidity after major trauma. Regulation of the
intestinal barrier is complex and involves the commensal microflora, a network of
immune cells and importantly, an integrated network of neuronal cells of the enteric
nervous system (ENS).
It is now well established that enteric neurons participate in nearly all
aspects of gastrointestinal tract (GI) function, including epithelial transport, motility,
mucosal immune defense, release of gut peptides from enteroendocrine cells, and
mucosal blood flow. Under normal conditions, enterocyte tight junction (TJ)
proteins allow the gut epithelium to function as a barrier thus regulating access for
luminal bacteria to mucosal phagocytes. Chronic stress or surgical trauma can alter
expression of TJ and adherence junction (or zonula adherens) proteins and
antimicrobial peptides allowing gut pathogens to adhere and cross the gut
epithelium. Such proteins are composed of transmembrane proteins occludin,
claudins and other junctional adherent molecules that are connected to the
cytoskeleton via a complex of multiple proteins. Changes in epithelial barrier
function and TJ expression also form the basis of defects in wound healing and/or
increased electrolyte secretion that are often seen in IBD, and that perpetuate disease
progression. Therefore, approaches aimed at protection or re-establishing epithelial
barrier functions could be of interest. In this context, the role of the ENS in the
- 131 -
Chapter 6
regulation of barrier function and the gut immune response is emerging. Recent
advances have highlighted the ENS as playing a key role in the control of barrier
functions and have indicated that alterations of the ENS could be directly associated
with the development of IBD and its associated symptoms. One example thereof is
that cholinergic activity has been shown to be involved in regulation of the mucosal
barrier function, enterocyte endocytosis, and intestinal epithelial permeability. In
addition, cholinergic signalling is implicated in intestinal permeability changes
observed after chronic stress. The ENS may also exert regulation of intestinal barrier
function via a network of enteric glia that interact with epithelial TJs, possibly via
release of transforming growth factor (TGF)- or other mediators.
Here, we review regulation of intestinal barrier function by the ENS, and
focus mainly on the first component of intestinal barrier function: maintenance of
intestinal epithelial integrity.
- 132 -
The enteric nervous system as regulator of intestinal epithelial barrier function
The epithelial barrier function in health and disease
Epithelial cell function and tight junction protein assembly
Barrier interfaces between the external environment and sterile stromal
compartments are found throughout the entire body such as in skin, kidneys, and
intestine. However it has become increasingly clear that despite the presence of TJs
in all epithelia, the electrical resistance of the paracellular pathway varies more than
a 1000-fold between different epithelia. Hence, TJs in various tissues can have very
different barrier properties. The critical nature of a cellular barrier, and
consequences of barrier loss for immune homeostasis, can be demonstrated in burn
patients, in whom prognosis is directly related to the surface area damaged. Burns
represent a gross form of barrier loss, where there is direct damage to the epithelial
cells and exposure of underlying tissues. This damage results in a markedly
increased risk of infection.
The intestinal epithelial barrier (IEB) stays in close contact with the
environment and is composed of a monolayer of specialised intestinal epithelial cells
(IECs). The IEB has developed specific mechanisms to prevent access of luminal
contents into the lamina propria, which includes the restriction of paracellular
transport and the maintenance of the architecture of the epithelial barrier. This
function is brought about by the apical junctional complex, which is composed of
the TJ, or zonula occludens (ZO) and the subjacent adherens junction, or zonula
adherens. The desmosomes, or macula adherens, are located along the lateral
membranes beneath the adherens junction. Intracellular junctions including adherens
junctions, desmosomes, gap junctions and TJs tightly regulate paracellular transport
in conjunction. Whereas TJs seal the paracellular pathway, the adherens junctions
and desmosomes provide the strong bonds necessary to maintain cellular proximity
and allow TJ assembly. Adherens junctions are also critical for epithelial
polarisation and differentiation, mucosal morphogenesis, and tumour suppression,
- 133 -
Chapter 6
processes that occur through a variety of interactions with other proteins, including
actin and -catenin.
TJs are the most apical components of these intercellular junctions. The
main functions of TJs are to prevent diffusion of membrane proteins and lipids
between basolateral and apical membranes so that cell polarity is preserved (fence
function) and importantly, function as a selective barrier to paracellular transport
(barrier function). TJs complexes are composed of a network of proteins that are
coupled to actin filaments of the cytoskeleton 1. Occludin (62-82kDa), several
members of the claudin family (20-27kDa) and junctional adhesion molecule
(JAM)-1 (36-41kDa) are proteins that make up the membrane part of TJs
2-5
(see
figure 1). Occludin has to be phosphorylated to become integrated into the
membrane, whereas dephosphorylation directs occludin to intracellular pools
1,6-8
.
However, evidence from occludin-deficient mice indicates that occludin itself may
be dispensable as these mice are still able to form well-developed TJ strands and
retain normal IEB integrity
9,10
. Occludin seems to be more important in cell
signalling than in maintaining the IEB
9-11
. Probably, claudins are the main barrier-
forming proteins since different types of claudins are expressed in a restricted
number of cell types or during development. It has been assumed that claudin-2
forms mainly aqueous channels, whereas the permeability of macromolecules is
unaffected
2,9,10
. Overexpression of claudin-1 and -4, however, results in an
enhanced barrier function, indicating that these proteins play an important role in the
TJ complex formation
12,13
. Members of the ZO family are proteins that form a
bridge between these membrane proteins and actin filaments, which are connected to
the peri-junctional ring, a component of the cellular cytoskeleton 14,15.
Inflammatory mediators affecting epithelial TJ integrity
Increased concentrations of pro-inflammatory cytokines are present in the intestine
during the active phase of IBD but also in conditions such as haemorrhagic shock or
sepsis. In vitro studies in intestinal cell lines have demonstrated that pro-
- 134 -
The enteric nervous system as regulator of intestinal epithelial barrier function
inflammatory cytokines decrease the barrier function of intestinal epithelial
monolayers and induce reorganisation of several TJ-associated proteins, including
ZO-1, JAM-1, occludin and claudin-1, and -4
16,17
. Examples of such cytokines that
cause TJ rearrangements are TNF-, IFN-, IL-8 and IL-1
18-20
. These cytokines
influence the IEB primarily by acting on the epithelial expression and activation of
myosin light chain kinase (MLCK) through PKC-activation. Upon activation,
MLCK phosphorylates myosin light chain (MLC) which in turn causes contraction
of the perijunctional ring, a component of the cellular cytoskeleton, so that
permeability of TJs increased 21-23. IL-1 increases the intestinal permeability by the
induction of MLCK gene transcription resulting in an increased MLCK protein
activity, probably mediated by a rapid activation of transcription factor NF-B
24
.
IL-1 mediates increased intestinal permeability via increased paracellular transport
of luminal antigens 19. Also TNF--mediated increased intestinal permeability leads
to an NF-ĸB dependent down-regulation of ZO-1 proteins and alteration in
junctional localisation 25. In turn, the anti-inflammatory cytokines IL-10, TGF- and
IL-17 protect from loss of TJ proteins
19
. The role of IL-6 in modulation of the
epithelial barrier is controversial, and may depend on the specific cell type or model
system used 19 . IL-6, as well as IL-13
26
and TNF affect epithelial permeability and
cell turnover through activation of pro-apoptotic pathways
activation of PI-3kinase dependent signalling pathways
27
and possibly the
19
. Altogether, cytokine-
mediated barrier dysfunction remains poorly defined, and cytokines modulate TJs
through distinct mechanisms and intracellular signalling pathways. Data indicate
that ZO-1 might be an effector molecule in this process.
- 135 -
Chapter 6
Intestinal pathogens
During intestinal infections, enteric pathogens have developed several mechanisms
to disrupt the IEB, which clinically often results in diarrhoea. This occurs mainly by
modulating the perijunctional actomyosin ring or by interfering with TJ proteins
directly. TLR triggering on enterocytes intuitively provides a protective role in
epithelial integrity 28, which is especially evident for TLR2 and 4 activation 29. This
link between TLR2 and TJ proteins was shown to be mediated via Connexin-43,
during acute and chronic inflammatory injury of the intestinal epithelia. This
protective barrier regulatory function was even put forward to account for the
association of ulcerative colitis-associated TLR2-R753Q mutant genes 28,29.
Bacterial products degenerate or phosphorylate specific TJ proteins or use
them as a receptor so that these proteins become dysfunctional or are reorganised
and the efficiency of TJs decreases. Enteropathogenic Escherichia coli disrupts the
IEB by changing the distribution of ZO-1 and occludin from the membrane into the
cytosol in vivo
30
. Moreover, adherent-invasive E. coli increases intestinal
permeability by an increased macromolecular flux, a decreased TEER and
redistribution of ZO-1
. Besides E.coli, other intestinal pathogens disrupt the
31
integrity of the IEB, such as Salmonella, Campylobacter and Shigella. Salmonella
thyifimurium influences TJs by dephosphorylation of occludin and degradation of
ZO-1
32
. Campylobacter jejuni distributes occludin from an intercellular to an
intracellular location, decreases the TEER and increases the IL-8 production by
epithelial cells by activation of the NF-B pathway
33
. Probiotics seem to prevent
disruption of the epithelial barrier. Lactobaccilus plantarum has been found to
protect the integrity of the epithelial barrier in case of an enteroinvasive infection of
E. coli in vitro via the prevention of ZO-1, occludin, claudin-1 and JAM-1
rearrangement. Besides, Lactobacilli facilitate the maintenance of the epithelial
barrier during Shigella dysententeriae infection in rats 34. The same results are found
for Lactobaccilus rhamnosus which maintain the barrier function in case of an
enterohaemorrhagic E. coli infection
- 136 -
35
. Also, proteases released from luminal
The enteric nervous system as regulator of intestinal epithelial barrier function
bacteria mediate intestinal barrier through cleavage of protease-activated receptor
(PAR)-2. In IBD, the epithelial barrier is inflamed without obvious exogenous
factors like a bacterial or viral infection. Nevertheless, colonic biopsies from
Crohn’s disease (CD) patients contain decreased numbers of Lactobaccilus and
Bifiobacteria, whereas the mucosa and probably even the intraepithelial layer
contain an increased population of adherent bacteria
36,37
. Probably, the immune
system itself influences the composition of TJs by the production of proinflammatory cytokines. Patients with CD have increased levels of the pore-forming
claudin-2, whereas the sealing claudins 5 and 8 are downregulated
38
. In colonic
tissues of ulcerative colitis patients, the expression of ZO-1, JAM-1 and claudin-1 is
downregulated 39. Moreover, in colonic epithelium, the expression of ZO-1 is lost in
DSS-induced colitis in mice and also mice that are JAM deficient are more
susceptible for the development of colitis 40,41
Mast cell mediators effecting barrier function
Initially studied in the context of allergic diseases, we now know that mast cells are
highly versatile cells that not only have a sentinel role in host defence but also play a
central role in intestinal disorders like IBD and IBS. Mast cells can be activated by a
variety of stimuli and the type of stimulus determines their mediator release profile
and subsequent consequences for neighbouring cells. In reference to the long list of
cytokines that may be involved in decreased barrier function (i.e. IL-10, IL-6, IL-13,
TGF-β and TNF-α) it is important to notice that all of these can be expressed by
mast cells 42. Most of them are de novo synthesised upon mast cell activation but an
important exception to this is TNF-α. Results obtained by Bischoff et al. not only
showed that this pro-inflammatory cytokine is constitutively expressed by these cells
but also indicated that approximately 60% of TNF-α positive cells in the gut are in
fact mast cells 43. As mentioned before, TNF-α induced barrier dysfunction depends
on MLCK-mediated modulation of TJs 25. In addition, however, it was also shown
that TNF-α potentiates histamine-induced ion secretion in enterocyte cell lines and
isolated distal colon
44
. Thus, although histamine is one of the classic mast cell
- 137 -
Chapter 6
mediators involved in itch, here it was shown that its synergistic action with TNF-α
induces enhanced chloride secretion across the intestinal epithelium. This is highly
relevant because it may lead to excessive water secretion and subsequent diarrhoea
as observed in e.g. IBD and IBS. Next to histamine, another preformed mediator
relevant to barrier dysfunction is tryptase which controls paracellular permeability
through PAR-2. Tryptase-mediated cleavage of the N-terminal extracellular domain
of this G-protein coupled receptor not only induces the redistribution of TJ proteins
via extracellular signal-related kinases (ERK1/2)
mediated activation of MLCK
45
but also via Ca2+/calmodulin
46
. Being far from complete, this small overview
clearly shows that mast cells and their mediators are major players in direct
modulation of intestinal barrier function.
- 138 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Figure 1. Model scheme of neural networks and epithelial integrity in the intestine. An integrated
network of intrinsic and extrinsic neurons mediate epithelial barrier function. A) Extrinsic and
intrinsic neuronal pathways may modulate neurotransmitter release in epithelia, possibly via
activation of enteric glial cells (green), or inflammatory cells known to be responsive
neurotransmitters, such as mast cells. B) Enterocytes may be in close contact to neurones, enteric
glial cells, mast cells and phagocytes. These cells interact with each other via the release of
neurotransmitters such as acetylcholine, but also (pro-inflammatory) cytokines including IL-1, also
produced by enteric glial cells. C) Epithelial tight junction integrity is known to be modulated via the
action of neurotransmitters directly, or inflammatory mediators. Tight junction rearrangement may be
affected via these mechanisms. Tight junctions are composed of transmembrane proteins including
JAM-1, occludin and claudins, which are connected to the actin skeleton via members of the zonula
occludens (ZO) family, MUPP1, MAG1 and cingulin.
- 139 -
Chapter 6
The enteric nervous system in intestinal barrier function
The central nervous system (CNS) interacts with the GI tract through the brain-gut
axis communicating in a bidirectional fashion largely through the enteric nervous
system (ENS) 47. The autonomic ENS comprises parasympathetic and sympathetic
systems that and can operate without the participation of the CNS, although the ENS
receives input directly from the brain through parasympathetic nerves (i.e. the vagus
nerve). The ENS is organised in several plexuses throughout the intestinal wall: the
myenteric (or Auerbach’s plexus, between circular and longitudinal muscle layer)
and submucosal (or Meissner’s plexus, in the submocosa) plexuses. The mucosal
layer also contains nerve networks known as the mucosal plexus, which contains
nerve endings that are in close contact with mucosal immune cells and enterocytes
(see figure 1). The ENS contains sensory neurons, interneurons, motor neurons,
which primarily control peristalsis, local changes in blood flow, and secretion of
water and electrolytes 48. An important component of the ENS are the enteric glial
cells (EGC), which form a large and widespread network at all levels of the GI tract
49
, and serve as intermediaries in the enteric neurotransmission and information
processing. They show morphologic and functional similarity to brain astrocytes and
control several aspects of gut function, including motility, microvascular circulation,
epithelial secretion of fluid, ions and bioactive peptides and intestinal barrier
function 50.
More than 30 different neurotransmitters exist in the ENS, with most
neurons expressing multiple transmitters 48. Like neurons of the CNS, ENS neurons
secrete acetylcholine (Ach) and a large number of other neurotransmitters and
neuropeptides including (adenosine triphosphate) ATP, NO, vasoactive intestinal
peptide (VIP), tachykinins (TK), Calcitonin gene related peptide (CGRP) and
neuropeptide Y (NPY) and Substance P (SP) reviewed elsewhere,47,48.
There are many indications that the GI tract’s specialised immune system
intimately interacts with the neuronal mediators released from the ENS. Many cells
- 140 -
The enteric nervous system as regulator of intestinal epithelial barrier function
of the Gut-associated Lymphoid tissue (GALT) produce for instance Ach and a wide
range of neuropeptides as well as the respective neuropeptides receptors 50. Besides
the well-known role of enteric neurons in controlling electrolyte secretion and
absorption, they also play a role in the control of intestinal paracellular permeability
and cell proliferation. Another cell population that may contribute to this neuroimmune axis in the gut are endocrine cells (EEC), which main function is to detect
changes in luminal nutrient content and signal to sensory afferents of the ENS. EEC
regulate satiety, motility and pH, or fluids, electrolytes and digestive enzymes via
reflex mechanisms. Mediators secreted by EEC include serotonin (5-HT), motilin,
glucagon-like-peptide (GLP), neuropeptide Y (NPY) and SP which renders these
cells potent activators of afferents of the sympathetic and parasympathetic nervous
systems.
Enteric glial cells as intermediates of neuronal barrier regulations
Similar to the role of astrocytes in maintaining the blood brain barrier, enteric glia
play an important role in regulating intestinal permeability. The importance of
enteric glia was noted in studies using targeted ablation of EGCs in transgenic mice.
Loss of EGCs was fatal in that animal model due to haemorrhagic necrosis of the
small intestine. Furthermore, in vitro data of co-culture models of enteric glia with
IECs lines have shown that glia decrease IEB permeability in part via the liberation
of a recently identified glial factor, GSNO (S-nitrosoglutathione), and the regulation
of ZO-1 and occludin expression. In 1998, Bush et al.
51
showed that EGCs act as
regulators of intestinal immunity. Ablation of ileal and jejunal glial cells in
transgenic mice resulted in a fatal intestinal inflammatory phenotype and
development of jejuno-ileitis similar to IBD
51,52
. In subsequent studies it was
demonstrated that EGCs are required to maintain mucosal barrier integrity
52
. In
cultured epithelial cells, enteric glia conditioned medium promoted intestinal barrier
properties and GNSO was identified as the crucial factor in maintaining barrier
homeostasis. GNSO-mediated regulation of mucosal barrier function was in part
influenced by altering expression of perijunctional F-actin and association of ZO-1
- 141 -
Chapter 6
and occludin with the actin-cytoskeleton
52
. In addition, i.p. GNSO injection
prevented barrier defects, cytokine induction and almost prevented enteritis caused
by ablation of enteric glia. In Ussing chambers, GNSO promoted intestinal barrier
function in biopsies from CD patients but not in healthy controls
. However, in
52
vitro, at low doses GNSO promotes epithelial barrier function, whereas at higher
doses GNSO that could be produced in pathological conditions disrupts epithelial
barrier integrity.
Although GNSO seems to be the main factor in barrier regulation by EGCs,
they also actively respond to inflammation and could be a significant source of
cytokines such as IL-1, TNF and IL-6
nitrergic neurotransmission
49
and also play a role in peptidergic and
53
. Moreover, EGCs were able to strongly inhibit IEC
proliferation in part by their release of TGF-β1
54
. The latter may explain the
observation that ablation of EGCs in vivo leads to an increased uptake of thymidine
in IECs and crypt hyperplasia. It is unclear if the role of glia in this network is
functionally pro- or anti-inflammatory 49. Therefore, enteric glia might alter barrier
function depending on the conditions of the local microenvironment. It is has
become clear however that the impact of EGCs on epithelia may be much more
elaborate, also indicated by a recent study in which it was demonstrated that EGCs
have a profound impact upon IEC transcriptome and induce a shift in IEC phenotype
towards increased cell adhesion and cell differentiation 55. In addition enteric glia are
neuroprotective in in vitro or ex vivo studies, through the factor GSH
56
.
Irrespectively, these data highlight the potential key role of EGC in the control of
IEB functions.
The vagus nerve and intestinal barrier function
Limiting intestinal barrier injury may play an important role in decreasing the
systemic inflammatory response associated with severe injury. It is generally
accepted that vagal activity can have a beneficial effect on the course of intestinal
inflammation, as shown for post-operative ileus, colitis, intestinal infection, septic
- 142 -
The enteric nervous system as regulator of intestinal epithelial barrier function
shock, and pancreatitis
57
. For instance, in a model of haemorrhagic shock, CCK-
dependent stimulation of vagal activity by high fat nutrition has been shown to
maintain intestinal barrier integrity. Translocation of bacteria, permeability to HRP,
disturbed expression of ZO-1
58
and enterocyte damage
59
in high-lipid-treated
animals was significantly reduced when compared with those in low-lipid-treated
and fasted controls. The protective role of vagal nerve stimulation (VNS) has been
described in several other studies. Vagal efferent activity has previously been shown
to decrease histological gut injury and intestinal inflammation in a model of colitis
60,61
. VNS has also been shown to ameliorate the severity of post-operative ileus in a
murine model 62, further supporting the potential of VNS to preserve the epithelial
barrier function and ameliorate herewith associated intestinal inflammatory disease.
Activation of vagal activity to preserve barrier function has been achieved via
58,59
, and electrical techniques
pharmacological, nutritional
62,63
. Alternatively, in
addition to fat feeding-mediated vagal improvement in barrier function in a shock
model, peripheral as well as intracerebroventricular injection with ghrelin
ameliorated the disrupted barrier function in rat models of intestinal ischemiareperfusion
64
and sepsis
64
. In both studies, vagotomy prevented the effect of
ghrelin, demonstrating that these effects are depended on an intact vagus nerve.
- 143 -
Chapter 6
A
Sham
VNS 5V
D
160
CFU/g MLN
120
80
40
*
Anaerobic
**
80
40
VNS 5V
VNS 5V
sham
0
sham
Carb 1M
Nico 1M
Veh
sham
0
Aerobic
120
VNS
HRP flux (pmol/h/cm2)
C
B
Figure 2. Vagal nerve stimulation and enhanced bacterial Translocation. A) and B) Translocation
of FITC-labelled E. Faecium (white dots) to the mucosal compartment of the ileum is increased in
vagal nerve stimulation (VNS)-treated mice (B) compared to control mice (sham) (A). E. faecium is
not taken up via lysosomes, but rather translocates via paracellular routes since LAMP-2 and E.
faecium do not co-localise. For colour pictures see page 215. C) Tissue mounted after VNS did not
show an increased HRP flux in Ussing chamber analyses; however HRP flux was increased by the
activation of acetylcholine receptors by 1M nicotine or 1M carbachol. D) Translocation of aerobic
and anaerobic bacteria to the mesenteric lymph nodes (MLN) is significantly increased (p<0.01 and
p<0.001) respectively) in VNS-treated mice compared to the control mice (sham). Hence the vagus
nerve activity may also target dendritic cell function. Tytgat Institute, Amsterdam, Unpublished data
2010.
- 144 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Mechanisms of vagus nerve activity and barrier function in the gut
It is interesting to speculate on the working mechanism behind the vagal potential to
affect barrier function. Vagal activity may lead to peripheral release of it’s principal
neurotransmitter Ach, but the vagus nerve particularly projects to other postganglionic enteric neurons, in particular VIP
serotonin
65
(studied in the duodenum) and
66
. In fact, some of the functional effects of vagus nerve stimulation are
counteracted by VIP antisera 67. The release of VIP by enteric neurones is shown to
inhibit IEC proliferation and to maintain epithelial barrier integrity, and the effect of
VIP on epithelial permeability is concomitant with a neural-induced increase in ZO1 mRNA and protein expression in intestinal epithelial cells
68
. Besides VIP, Ach
increases paracellular permeability in the healthy gut 63, setting the basis for a fine
'tuning' of the barrier permeability by the ENS, either with or without the
intermediate action of enteric glia (see figure 2 and 3). Irrespectively, innate immune
cells express a broad range of nAchRs and mAchRs 69, and peptidergic receptors70,
and are therefore target cells for regulation through the ENS. In vitro and in vivo
studies have raised interest in targeting neurotransmitter receptors on immune cells
to modulate intestinal disease and counteract barrier disruption. However, very little
data is available on the interactions that really occur between ENS neurotransmitters
and immune cells in the intestinal tract and is subject to further investigation.
Furthermore, a likely cellular target of vagal modulation of the intestine is
through the activation of enteric glia. In the respect Gulbransen et al., have recently
shown that enteric neurons cause activation of EGCs through release of the
neurotransmitter ATP
71
. It will be interesting to investigate whether vagal nerve
activity can likewise relay functional changes via activation of EGCs. This would
demonstrate a link between the vagus nerve and enteric glia, and gives insight into
possible mechanisms by which the CNS communicates with the ENS following gut
inflammation.
- 145 -
Chapter 6
MLCK
IL1
NO
lymphatics
ECG
PVN
DMV
NTS
vagus nerve activity
Brainstem
Nodose
ganglia
Spinal cord
Figure 3. Scheme of neurogenic influences on epithelial barrier function. The vagus nerve
influences the permeability of the epithelial barrier via mast cells and other immune cells. Besides,
the vagus nerve communicates with enteric glial cells (EGCs) through enteric neurons. Enteric glia
produce S-nitrosoglutathione, which plays an important role in the homeostasis of the epithelial
barrier. Nevertheless, enteric glia can respond to inflammation by the production of IL-1 leading
to an increased epithelial permeability and activation of immune cells. Translocation of luminal
antigens to the lamina propria and tissue damage result in TLR and danger-associated molecular
pattern activation (DAMPs- including the pattern recognition receptor Receptor for Advanced
Glycosylated Endproducts; RAGE) of immune cells followed by cytokine and chemokine release.
Other immune cells, but also neurons and EGCs can show response to the cytokines and
chemokines and besides the induction of inflammation, also anti-inflammatory pathways will be
activated.
- 146 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Epithelial barrier dysfunction as a contributor of intestinal disease
pathology, focus on stress
Causes of intestinal barrier dysfunction can be quite diverse and range from
intestinal infection to allergic food components, malnutrition, toxic chemicals,
NSAIDs and mechanical trauma. In addition, during recent years we have gained a
lot of knowledge on the possible role of psychological-stress related impairment in
barrier function. Stress was shown to induce loss of barrier integrity not only in
small bowel and colon but, more recently, also in oesophagus.72 Although the latter
may explain why stress is not only associated with IBD
73
and IBS
74
but also with
gastro-oesophageal reflux disease (GERD), focus of most research has been on the
intestines 75. In this part of the review we will cover its role in especially IBD and
IBS.
Stress-induced mast cell degranulation alters epithelial permeability
Mast cells seem to play an important role in IBD and IBS and especially in IBSrelated animal models it was convincingly shown that stress-induced mast cell
degranulation can induce disease phenotype. Stress leads to peripheral (next to
central) release of corticotrophin releasing hormone (CRH) and, subsequently,
CRH-mediated degranulation of peripheral mast cells 76. We discussed earlier how
mast cell-derived cytokines and preformed mediators can directly modulate barrier
integrity by TJ rearrangements. It was also shown that stress will lead to increased
(mast cell dependent) bacterial adherence and penetration into enterocytes associated
with mucosal inflammation
77
. Importantly, although initial stress-related mast cell
degranulation will depend on peripheral CRH, the subsequent flaw in barrier
function evidently leads to the uptake of bacterial antigens and may thus be selfsustaining. Antigen-mediated degranulation of mast cells can lead to further
breakdown of barrier function and induce a vicious circle
76
that will have to be
counteracted by interfering mechanisms. In this respect, degranulating mast cells can
also activate protective mechanisms by themselves. Goblet cell secreted mucins
- 147 -
Chapter 6
form a physical barrier that prevents the attachment of enteric pathogens to the
epithelium 78 and it is known that stress-induced mast cell degranulation can lead to
increased colonic mucin release 79. Further, as mentioned before, mast cell-mediated
histamine-induced epithelial chloride secretion can lead to excessive intestinal water
secretion. Histamine also signals to the ENS and activates a neural defence program
leading to orthograde power propulsions in the small and large intestine
80
. These
combined actions can lead to powerful expulsion of a threatening pathogen and may
thus be highly beneficial to the organism as a whole. On the other hand, such mast
cell-mediated mechanisms also seem to play a role in e.g. diarrhoea-predominant
IBS and IBD and one can speculate whether the observed diarrhoea is either
protective or detrimental (or both). In IBD diarrhoea may be initiated to expel
luminal content relevant to disease pathogenesis but it is also known that stress can
lead to disease relapse. IBS, being a functional disorder, is diagnosed by symptoms
alone and stress is an important trigger for symptoms. Although some reports
suggest subtle qualitative changes in colonic flora of IBS patients 81, we do not know
whether stress induced diarrhoea leads to improved intra-luminal bacterial make-up.
Thus, at present, stress-induced barrier dysfunction in these disorders is being
regarded as detrimental instead of beneficial.
Stress related barrier dysfunction in IBD
While inflammation within the gut wall can clearly influence the perception of
visceral sensation by the CNS, it is also apparent that signals from the brain to the
gut are crucial in modifying the course of IBD progression. This phenomenon is best
exemplified by the large body of evidence that suggests psychological stress and
anxiety can negatively impact on the initiation, development and recurrence of
inflammation. Numerous studies in patients with IBD implicate a relationship
between stress-related personality traits (e.g. obsessive-compulsive disorders) or
increased life stressors and disease exacerbation. Both the sympathetic and
parasympathetic nervous system has been implicated in relaying stress signals from
brain to gut
- 148 -
82
. As mentioned before, stress can lead to a (mast cell dependent)
The enteric nervous system as regulator of intestinal epithelial barrier function
decrease in IEB function through alterations in TJ permeability. This may lead to
increased influx of antigens and bacteria followed by recruitment of inflammatory
cells. Indeed, in some studies the adverse effects of stress on gut physiology were
shown to depend on the recruitment and activation of immune cells, most notably
mast cells and lymphocytes
83
. Mast cell degranulation and related barrier
dysfunction may thus play an important role in IBD, but relevant investigations were
not necessarily done in colitis models. In relation to this, it was shown that prior
exposure to stress significantly enhances experimental colitis. Surprisingly, this
stress-effect was associated with decreased release of protective mucins 84. Although
these data seemingly contradict earlier reports on stress-induced mast cell
degranulation and associated increase in mucin levels, this discrepancy may be
explained by the observed loss of goblet cells in the colitis model. Lastly, neuronal
stress signals can also negatively influence immune barrier function at another level.
IgA is continuously being secreted into the lumen to contain commensal bacteria
outside the epithelial barrier. Stress was shown to decrease IgA production in
colonic mucosa
85
and may thus promote bacterial adherence and result in local
immune activation, inflammation and perpetuation of ongoing disease state.
Stress related barrier dysfunction in IBS
Abdominal pain and motility changes are key features of IBS and acute stress is a
known trigger for these symptoms in patients and in animal models. Furthermore,
increased intestinal permeability is present in at least part of IBS patients
86
and,
although conflicting data exist, patients may have enhanced numbers of mucosal
mast cells. Even more importantly, the number of degranulated mast cells was
shown to be higher compared to controls 87. The latter is relevant because in jejunum
of IBS patients, acute stress was shown to induce release of mast cell mediators, and
this was associated with enhanced permeability to albumin
88
. These human data
seemingly confirm earlier observations in animal models: stress induces peripheral
mast cell degranulation and subsequent barrier dysfunction. In rats, Ait-Belgnaoui et
al. were able to show that stress-induced barrier dysfunction can be a direct cause
- 149 -
Chapter 6
for visceral hypersensitivity (enhanced sensitivity to a normal stimulus) 89. Pre-stress
treatment with a TJ blocker or MLCK inhibitor preserved barrier integrity and
prevented the development of enhanced sensitivity to colonic distension. Because
visceral hypersensitivity is considered a pathophysiological mechanism in IBS, these
results suggest that stress-induced impairments in barrier function can be a main
trigger to initiate and/or perpetuate symptoms in this disorder. Despite these
evidences, a cautionary note is needed because most data on stress-related
mechanisms in relation to IBS were indeed gathered in animal models. Further,
although most data seem to point towards a central role for mast cells other
mechanisms may also be involved. Results obtained by Demaude et al. suggested
that early post-stress epithelial barrier defects do not depend on mast cell mediators
but on pancreatic trypsin instead 90.
- 150 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Vagal modulation via release of
neurotransmitters
Epithelial permeability
Proteases1
TryptasePAR-2 cleavage2
Mast cells
tight junction rearrangement 3,4
Histamine
De novo synthesis:
Pro-inflammatory cytokines
(i.e. IL-1beta, TNF)
Mast cells produce several factors that
increase the epithelial permeability.
Proteases and tryptase rearrange tigth
junction via the cleavage of PAR-2. Also
histamine and pro-inflammatory cytokines
including IL-1 and TNF- increase the
intestinal epithelial permeability.
1.
2.
3.
4.
Santos, J. et al., Gut 2001
Wallon, C. et al., Gut 2008
Jacob, C. et al., JBC 2005
Bueno, L. et al. Neurogastroenterol Motil 2008
Figure 4. Potential mast cell regulation of the epithelial barrier. Stress-induced mast cell
degranulation results in the production of several factors that increase the epithelial permeability.
Proteases and tryptase rearrange tight junctions via the cleavage of Protease-activated receptors
(PARs) resulting in leakage of the epithelial barrier. Also histamine and pro-inflammatory cytokines
including IL-1 and TNF- increase the intestinal epithelial permeability so that luminal antigens
have access to the lamina propria.
- 151 -
Chapter 6
Epithelial integrity in intestinal disease
Altered intestinal epithelial permeability is not only associated with major trauma
and septic states, but also perpetuates intestinal disease including IBD, stress
induced conditions such as IBS and food allergy, and intestinal infections 91. During
these pathological disorders, mechanisms such as TJ rearrangements, decreased
mitochondrial function and changes in apoptosis and proliferation
26
of the
epithelium all might contribute to barrier disruption.
Inflammatory Bowel Disease
Clearly, in chronic inflammatory settings alterations in the ENS that contribute to
the enhanced permeability were observed
47
. These alterations include a change in
number and size of glial cells and enteric neuronal cell bodies and their nerve fibres,
and remodelling of neural synapses. Also, changes in chemical coding of the
neurons occur, with alterations in VIP and NO-synthase NPY, PACAP, CHAT and
also altered receptor expression 47. These changes occur in inflamed areas but also at
remote sites and are probably responsible for the widespread nature of changed
physiology in IBD 92. Furthermore, altered motility was observed at a remote noninflamed site and at inflamed sites in ulcerative colitis patients or animal models of
IBD 93. These findings suggest that the ENS may play a role not only during active
inflammation, but also in the development of disease. This is illustrated by the fact
that ENS disturbances will predispose to postoperative recurrence and stress induced
relapses. Healthy, first-degree relatives of patients with IBD were reported to have
increased intestinal permeability and altered intestinal permeability is predictive of
clinical relapse in patients with clinically inactive IBD 91. However, ENS alterations
are mostly secondary and whether changes in ENS precede gut diseases needs to be
confirmed.
- 152 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Barrier dysfunction in functional bowel disease
Intestinal ischemia occurs in several conditions including (non) abdominal surgery,
major trauma, cardiac arrest and is responsible for the barrier disturbances and
bacterial translocation to distant organs. Translocation of bacteria or endotoxins, is
an important mediator in the pathogenesis of sepsis during these conditions.
Ischemic conditions in the intestine result in selective intestinal actin cytoskeleton
and TJ disruption, as has been shown in a model of haemorrhagic shock 94. In animal
models of intestinal ischemia-reperfusion, the enzyme xanthine oxidase (XO) is
activated, inducing oxidative stress
95
that is the cause of morphological changes of
the epithelial monolayer and alterations of enterocyte mitochondrial function
96,97
.
98
Mitochondrial dysfunction might also induce transcellular uptake of bacteria , but
this process is not well understood thus far. Also, exaggerated local and systemic
immune responses that occur during ischemic conditions might further contribute to
barrier dysfunction 59,99.
Septic ileus
The occurrence of septic morbidity and even multiple organ failure in serious
conditions such as surgery, trauma, ischemia reperfusion injury might be the result
of a breakdown of the intestinal barrier and subsequent bacterial translocation100. In
a recent study including 927 patients over 13 years showed that bacterial
translocation was associated with increased postoperative septic morbidity in
surgical patients 101. However, the evidence for the so-called ‘gut origin of sepsis’ is
at least in humans, controversial
102
. The development of septic morbidity is
multifactorial and in certain patients measures taken to prevent septic morbidity
such as selective gut decontamination, the use of pre- or probiotics have not been
successful.
- 153 -
Chapter 6
Post-operative Ileus
The occurrence of barrier dysfunction following abdominal surgery may also
contribute to the pathogenesis of gastrointestinal stasis and post-operative or septic
ileus. Although gastric and colonic dysmotility are regarded as important
contributors to ileus, small bowel motility is an important factor in the regulation of
the enteric bacterial population. A delayed transit time results in bacterial
overgrowth, especially in the small bowel, and predisposes to bacterial translocation
103
. Bacteria from the intestinal lumen might activate intestinal resident
macrophages, given the observation that pre-treatment with antibiotics prior to the
intestinal manipulation reduced inflammatory responses. The ileus-associated
bacterial translocation reflects disturbances in intestinal barrier function and does
not only refer to the transepithelial passage of viable bacteria, but also (endo) toxins
or antigens from the intestinal lumen. This is illustrated by the observation that in
abdominal surgery, barrier dysfunction has been associated with increased
postoperative septic morbidity in surgical patients undergoing laparotomy.
Moreover, in an experimental model for post-operative ileus the occurrence of ileus
and the reduction of small intestinal smooth muscle contractility were not observed
after surgery in TLR4-deficient mice
104
leading to post-operative ileus in figure 5.
- 154 -
. See the proposed sequence of events
The enteric nervous system as regulator of intestinal epithelial barrier function
Intestinal manipulation (IM)
Impaired gastro-intestinal
motility
Mast cell
permeability
Post operative Ileus
Inflammatory infiltrate
Figure 5. Neurogenic influences on epithelial barrier function, contributing to pathogenesis of
POI. Intestinal manipulation (IM) results in an impaired gastro-intestinal motility leading to postoperative ileus (POI). IM leads to degradation of mast cells, which increases the permeability of the
intestinal epithelial barrier so that luminal antigens have access to the lamina propria. Residential
macrophages between the circular and longitudinal muscle layers become activated by mast cells and
luminal antigens, resulting in the recruitment of other immune cells. These inflammatory processes
activate inhibitory neuronal pathways, which will lead to a general impairment of the gastrointestinal
motility.
- 155 -
Chapter 6
In human subjects, it has been shown that gut barrier loss in children
undergoing major non-abdominal surgery is preceded by hypotension, mesenterial
hypoperfusion and enterocyte damage
105
. As indicated above, similar results were
obtained from animal studies where surgery induces production of free oxygen
radicals resulting in disturbances in enterocyte proliferation and subsequent
reduction in epithelial barrier function. In conjunction, we have now unpublished
data in which we observed that arterial blood pressure measured in the carotid artery
is greatly reduced during manipulation of the intestine during surgery, and that this
drop in blood pressure remains until the animals recover from anaesthesia. Most
likely, blood pressure in the carotid artery reflects the blood pressure of the internal
organs, but we cannot exclude that perfusion in the small intestine is different from
the carotid artery.
- 156 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Prospect of modulation of barrier function as a treatment in intestinal
inflammatory disease; 5 year view
Clearly, the expression of several TJ proteins is affected in IBD patients. Patients
with CD have increased levels of the pore-forming claudin-2, whereas the sealing
claudins 5 and 8 are downregulated. In colonic tissues of ulcerative colitis patients,
the expression of ZO-1, JAM-1 and claudin-1 is downregulated. Moreover, in
colonic epithelium, the expression of ZO-1 is lost in DSS-induced colitis in mice
and also mice that are JAM deficient are more susceptible for the development of
colitis
40,41
. The decrease in epithelial barrier contributes further to inflammatory
reactions during IBD but until now, little evidence suggests that it might be a
primary causative factor predisposing to disease development
91
. A considerable
body of evidence from animal models and patients, dated as long as 50 years ago,
has emerged that demonstrate that dramatic alterations occur in the ENS under
conditions such as chronic inflammation during IBD (though more pronounced in
ulcerative colitis). Such changes can be apparent as gross changes in the
morphology and architecture of neural ganglia and nerve cell bodies or can be seen
as subtle changes in the expression of neurotransmitters or their receptors at
synapses within the gut wall. Changes that occur in the ENS are clearly coupled to
alterations in gut physiology and permeability and are likely to play an important
role in the pathogenesis of the disease.
Treatment with chemical messengers could restore ENS function and
thereby the ongoing inflammation and could also prevent relapses at remote sites.
Indeed, administration of neuropeptides including VIP, NPY, cortistatin, opioids,
and NK-1R and CRHR antagonists ameliorated disease in IBD animal models. Also,
cholinergic agonists might have beneficial effects. In some studies with
experimental animal models of intestinal inflammation, animals were treated with
cholinergic agonists such as nicotine but with conflicting results. In ulcerative colitis
patients, nicotine patches have shown to be effective to reduce inflammation, but
due to side-effects this treatment is not applicable in patients. Therefore, a more
- 157 -
Chapter 6
selective treatment might be desirable to make use of the cholinergic antiinflammatory pathway. In one study, enhancement of sympathetic activity in
ulcerative colitis was shown
106
and treatment with an -adrenergic clonidine
reduced sympathetic overactivity and was associated with clinical and endoscopical
improvement as compared to placebo. The use of neuronal-derived messengers to
relieve bowel disease might come from the field of psychopathologies such as
anxiety and depression. The first clinical report that a CRHR1 antagonist (R121919)
significantly reduces depression and anxiety scores in patients with major
depression, also assigns therapeutic value to CRHR1 antagonists in the treatment of
IBS patients with stress-related symptom generation 107.
Taken together, novel therapeutic approaches designed to specifically target
and disrupt neuro-immune communication in IBD would be likely to benefit in
treatment of disease. The advantages of the use of neuron-derived chemical
messengers are that they are short lived and act local. Importantly, in addition to the
potential to ameliorate IEB, neuronal messengers also affect inflammatory
processes, motility, mucus production and water and electrolyte secretion, thereby
further contributing to disease amelioration.
- 158 -
The enteric nervous system as regulator of intestinal epithelial barrier function
Acknowledgements and grant support
All authors concur with the submission and the material submitted for publication
has not been previously reported and is not under consideration for publication
elsewhere. The authors state that there is no conflicting financial interest.
The authors gratefully acknowledge grant support from Top Institute Pharma (SAS),
7th European Framework Program (iPODD to RvdW) and the 6th European
Community Framework Programme Marie Curie (WdJ), Dutch Organisation for
Scientific Researc (NWO VIDI grant, to WdJ and NWO VICI grant, to GEB and
MIV).
Mrs I.E. Kos-Oosterling (Academic Medical Centre, Amsterdam) is gratefully
acknowledged for providing illustrations.
- 159 -
Chapter 6
References
1 Tsukita,S. et al. (2001) Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2, 285-293
2 Furuse,M. et al. (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell
Biol. 123, 1777-1788
3 Furuse,M. et al. (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions
with no sequence similarity to occludin. J. Cell Biol. 141, 1539-1550
4 Heiskala,M. et al. (2001) The roles of claudin superfamily proteins in paracellular transport. Traffic. 2,
93-98
5 Martin-Padura,I. et al. (1998) Junctional adhesion molecule, a novel member of the immunoglobulin
superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol.
142, 117-127
6 Andreeva,A.Y. et al. (2001) Protein kinase C regulates the phosphorylation and cellular localization of
occludin. J. Biol. Chem. 276, 38480-38486
7 Clarke,H. et al. (2000) Protein kinase C activation leads to dephosphorylation of occludin and tight
junction permeability increase in LLC-PK1 epithelial cell sheets. J. Cell Sci. 113 ( Pt 18), 3187-3196
8 Sakakibara,A. et al. (1997) Possible involvement of phosphorylation of occludin in tight junction
formation. J. Cell Biol. 137, 1393-1401
9 Saitou,M. et al. (1998) Occludin-deficient embryonic stem cells can differentiate into polarized
epithelial cells bearing tight junctions. J. Cell Biol. 141, 397-408
10 Saitou,M. et al. (2000) Complex phenotype of mice lacking occludin, a component of tight junction
strands. Mol. Biol. Cell 11, 4131-4142
11 Barrios-Rodiles,M. et al. (2005) High-throughput mapping of a dynamic signaling network in
mammalian cells. Science 307, 1621-1625
12 Inai,T. et al. (1999) Claudin-1 contributes to the epithelial barrier function in MDCK cells. Eur. J. Cell
Biol. 78, 849-855
13 Van,I.C. et al. (2001) Regulated expression of claudin-4 decreases paracellular conductance through a
selective decrease in sodium permeability. J. Clin. Invest 107, 1319-1327
14 Fanning,A.S. et al. (1998) The tight junction protein ZO-1 establishes a link between the
transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 273, 29745-29753
15 Itoh,M. et al. (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO3, with the COOH termini of claudins. J. Cell Biol. 147, 1351-1363
16 Bruewer,M. et al. (2003) Proinflammatory cytokines disrupt epithelial barrier function by apoptosisindependent mechanisms. J. Immunol. 171, 6164-6172
17 Zolotarevsky,Y. et al. (2002) A membrane-permeant peptide that inhibits MLC kinase restores barrier
function in in vitro models of intestinal disease. Gastroenterology 123, 163-172
- 160 -
The enteric nervous system as regulator of intestinal epithelial barrier function
18 Al-Sadi,R. et al. (2008) Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction
permeability. J. Immunol. 180, 5653-5661
19 Al-Sadi,R. et al. (2009) Mechanism of cytokine modulation of epithelial tight junction barrier. Front
Biosci. 14, 2765-2778
20 Boivin,M.A. et al. (2009) Mechanism of interferon-gamma-induced increase in T84 intestinal
epithelial tight junction. J. Interferon Cytokine Res. 29, 45-54
21 Anderson,J.M. and Van Itallie,C.M. (1995) Tight junctions and the molecular basis for regulation of
paracellular permeability. Am. J. Physiol 269, G467-G475
22 Hecht,G. et al. (1996) Expression of the catalytic domain of myosin light chain kinase increases
paracellular permeability. Am. J. Physiol 271, C1678-C1684
23 Turner,J.R. (2009) Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9,
799-809
24 Al-Sadi,R.M. and Ma,T.Y. (2007) IL-1beta causes an increase in intestinal epithelial tight junction
permeability. J. Immunol. 178, 4641-4649
25 Ma,T.Y. et al. (2005) Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight
junction barrier: role of myosin light-chain kinase protein expression. Am. J. Physiol Gastrointest. Liver
Physiol 288, G422-G430
26 Schulzke,J.D. et al. (2006) Disrupted barrier function through epithelial cell apoptosis. Ann. N. Y.
Acad. Sci. 1072, 288-299
27 Jin,X. et al. (2009) Interleukin-6 Is an Important In Vivo Inhibitor of Intestinal Epithelial Cell Death in
Mice. Gut
28 Cario,E. (2008) Innate immune signalling at intestinal mucosal surfaces: a fine line between host
protection and destruction. Curr. Opin. Gastroenterol. 24, 725-732
29 Ey,B. et al. (2009) TLR2 mediates gap junctional intercellular communication through connexin-43 in
intestinal epithelial barrier injury. J. Biol. Chem. 284, 22332-22343
30 Zhang,Q. et al. (2009) Enteropathogenic Escherichia coli changes distribution of occludin and ZO-1 in
tight junction membrane microdomains in vivo. Microb. Pathog.
31 Wine,E. et al. (2009) Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional
complexes in polarized epithelia. BMC. Microbiol. 9, 180
32 Kohler,H. et al. (2007) Salmonella enterica serovar Typhimurium regulates intercellular junction
proteins and facilitates transepithelial neutrophil and bacterial passage. Am. J. Physiol Gastrointest. Liver
Physiol 293, G178-G187
33 Chen,M.L. et al. (2006) Disruption of tight junctions and induction of proinflammatory cytokine
responses in colonic epithelial cells by Campylobacter jejuni. Infect. Immun. 74, 6581-6589
34 Moorthy,G. et al. (2009) Lactobacilli facilitate maintenance of intestinal membrane integrity during
Shigella dysenteriae 1 infection in rats. Nutrition 25, 350-358
- 161 -
Chapter 6
35 Johnson-Henry,K.C. et al. (2008) Lactobacillus rhamnosus strain GG prevents enterohemorrhagic
Escherichia coli O157:H7-induced changes in epithelial barrier function. Infect. Immun. 76, 1340-1348
36 Favier,C. et al. (1997) Fecal beta-D-galactosidase production and Bifidobacteria are decreased in
Crohn's disease. Dig. Dis. Sci. 42, 817-822
37 Swidsinski,A. et al. (2002) Mucosal flora in inflammatory bowel disease. Gastroenterology 122, 4454
38 Zeissig,S. et al. (2007) Changes in expression and distribution of claudin 2, 5 and 8 lead to
discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut 56, 61-72
39 Kucharzik,T. et al. (2001) Neutrophil transmigration in inflammatory bowel disease is associated with
differential expression of epithelial intercellular junction proteins. Am. J. Pathol. 159, 2001-2009
40 Laukoetter,M.G. et al. (2007) JAM-A regulates permeability and inflammation in the intestine in vivo.
J. Exp. Med. 204, 3067-3076
41 Vetrano,S. et al. (2008) Unique role of junctional adhesion molecule-a in maintaining mucosal
homeostasis in inflammatory bowel disease. Gastroenterology 135, 173-184
42 Marshall,J.S. (2004) Mast-cell responses to pathogens. Nat. Rev. Immunol. 4, 787-799
43 Bischoff,S.C. et al. (1999) Mast cells are an important cellular source of tumour necrosis factor alpha
in human intestinal tissue. Gut 44, 643-652
44 Oprins,J.C. et al. (2002) Tumour necrosis factor alpha potentiates ion secretion induced by histamine
in a human intestinal epithelial cell line and in mouse colon: involvement of the phospholipase D
pathway. Gut 50, 314-321
45 Jacob,C. et al. (2005) Mast cell tryptase controls paracellular permeability of the intestine. Role of
protease-activated receptor 2 and beta-arrestins. J. Biol. Chem. 280, 31936-31948
46 Cenac,N. et al. (2004) PAR2 activation alters colonic paracellular permeability in mice via IFNgamma-dependent and -independent pathways. J. Physiol 558, 913-925
47 Taylor,C.T. and Keely,S.J. (2007) The autonomic nervous system and inflammatory bowel disease.
Auton. Neurosci. 133, 104-114
48 Furness,J.B. (2000) Types of neurons in the enteric nervous system. J. Auton. Nerv. Syst. 81, 87-96
49 Ruhl,A. (2005) Glial cells in the gut. Neurogastroenterol. Motil. 17, 777-790
50 Genton,L. and Kudsk,K.A. (2003) Interactions between the enteric nervous system and the immune
system: role of neuropeptides and nutrition. Am. J. Surg. 186, 253-258
51 Bush,T.G. et al. (1998) Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic
mice. Cell 93, 189-201
52 Savidge,T.C. et al. (2007) Enteric glia regulate intestinal barrier function and inflammation via release
of S-nitrosoglutathione. Gastroenterology 132, 1344-1358
- 162 -
The enteric nervous system as regulator of intestinal epithelial barrier function
53 Aube,A.C. et al. (2006) Changes in enteric neurone phenotype and intestinal functions in a transgenic
mouse model of enteric glia disruption. Gut 55, 630-637
54 Neunlist,M. et al. (2007) Enteric glia inhibit intestinal epithelial cell proliferation partly through a
TGF-beta1-dependent pathway. Am. J. Physiol Gastrointest. Liver Physiol 292, G231-G241
55 Van,L.L. et al. (2009) Regulation of intestinal epithelial cells transcriptome by enteric glial cells:
impact on intestinal epithelial barrier functions. BMC. Genomics 10, 507
56 Abdo,H. et al. (2009) Enteric glial cells protect neurons from oxidative stress in part via reduced
glutathione. FASEB J.
57 Van Der Zanden,E.P. et al. (2009) The vagus nerve as a modulator of intestinal inflammation.
Neurogastroenterol. Motil. 21, 6-17
58 Luyer,M.D. et al. (2005) Nutritional stimulation of cholecystokinin receptors inhibits inflammation via
the vagus nerve. J. Exp. Med. 202, 1023-1029
59 de Haan,J.J. et al. (2008) Postshock intervention with high-lipid enteral nutrition reduces inflammation
and tissue damage. Ann. Surg. 248, 842-848
60 Ghia,J.E. et al. (2006) The vagus nerve: a tonic inhibitory influence associated with inflammatory
bowel disease in a murine model. Gastroenterology 131, 1122-1130
61 Ghia,J.E. et al. (2007) The protective effect of the vagus nerve in a murine model of chronic relapsing
colitis. Am. J. Physiol Gastrointest. Liver Physiol 293, G711-G718
62 Boeckxstaens,G.E. and de Jonge,W.J. (2009) Neuroimmune mechanisms in postoperative ileus. Gut
58, 1300-1311
63 Van Der Zanden,E.P. et al. (2009) Vagus nerve activity augments intestinal macrophage phagocytosis
via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology 137, 1029-39, 1039
64 Wu,R. et al. (2009) Orexigenic hormone ghrelin ameliorates gut barrier dysfunction in sepsis in rats.
Crit Care Med. 37, 2421-2426
65 Yuan,P.Q. et al. (2005) Central vagal stimulation activates enteric cholinergic neurons in the stomach
and VIP neurons in the duodenum in conscious rats. Peptides 26, 653-664
66 Kirchgessner,A.L. and Gershon,M.D. (1989) Identification of vagal efferent fibers and putative target
neurons in the enteric nervous system of the rat. J. Comp Neurol. 285, 38-53
67 Dahlstrand,C. et al. (1989) VIP antisera inhibit the relaxatory motor responses of the feline sphincter
of Oddi and gall-bladder induced by VIP or vagal nerve stimulation. Acta Physiol Scand. 137, 375-378
68 Neunlist,M. et al. (2003) Human ENS regulates the intestinal epithelial barrier permeability and a tight
junction-associated protein ZO-1 via VIPergic pathways. Am. J. Physiol Gastrointest. Liver Physiol 285,
G1028-G1036
69 Wessler,I. and Kirkpatrick,C.J. (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic
system in humans. Br. J. Pharmacol. 154, 1558-1571
- 163 -
Chapter 6
70 Snoek,S.A. et al. (2009) Neuropeptide Receptors in Intestinal Disease: Physiology and Therapeutic
Potential. Curr. Pharm. Des
71 Gulbransen,B.D. and Sharkey,K.A. (2009) Purinergic neuron-to-glia signaling in the enteric nervous
system. Gastroenterology 136, 1349-1358
72 Farre,R. et al. (2007) Critical role of stress in increased oesophageal mucosa permeability and dilated
intercellular spaces. Gut 56, 1191-1197
73 Mawdsley,J.E. and Rampton,D.S. (2006) The role of psychological stress in inflammatory bowel
disease. Neuroimmunomodulation. 13, 327-336
74 Tache,Y. et al. (2009) A role for corticotropin-releasing factor in functional gastrointestinal disorders.
Curr. Gastroenterol. Rep. 11, 270-277
75 Gareau,M.G. et al. (2008) Pathophysiological mechanisms of stress-induced intestinal damage. Curr.
Mol. Med. 8, 274-281
76 van den Wijngaard,R.M. et al. (2010) Peripheral relays in stress-induced activation of visceral
afferents in the gut. Auton. Neurosci. 153, 99-105
77 Soderholm,J.D. et al. (2002) Chronic stress induces mast cell-dependent bacterial adherence and
initiates mucosal inflammation in rat intestine. Gastroenterology 123, 1099-1108
78 Lievin-Le,M., V and Servin,A.L. (2006) The front line of enteric host defense against unwelcome
intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin. Microbiol.
Rev. 19, 315-337
79 Castagliuolo,I. et al. (1998) Colonic mucin release in response to immobilization stress is mast cell
dependent. Am. J. Physiol 274, G1094-G1100
80 Wood,J.D. (2006) Histamine, mast cells, and the enteric nervous system in the irritable bowel
syndrome, enteritis, and food allergies. Gut 55, 445-447
81 Quigley,E.M. (2007) Bacterial flora in irritable bowel syndrome: role in pathophysiology, implications
for management. J. Dig. Dis. 8, 2-7
82 Saunders,P.R. et al. (2006) Noradrenergic and cholinergic neural pathways mediate stress-induced
reactivation of colitis in the rat. Auton. Neurosci. 124, 56-68
83 Farhadi,A. et al. (2007) Mucosal mast cells are pivotal elements in inflammatory bowel disease that
connect the dots: stress, intestinal hyperpermeability and inflammation. World J. Gastroenterol. 13, 30273030
84 Pfeiffer,C.J. et al. (2001) Reduction of colonic mucus by repeated short-term stress enhances
experimental colitis in rats. J. Physiol Paris 95, 81-87
85 Ponferrada,A. et al. (2007) The role of PPARgamma on restoration of colonic homeostasis after
experimental stress-induced inflammation and dysfunction. Gastroenterology 132, 1791-1803
86 Piche,T. et al. (2009) Impaired intestinal barrier integrity in the colon of patients with irritable bowel
syndrome: involvement of soluble mediators. Gut 58, 196-201
- 164 -
The enteric nervous system as regulator of intestinal epithelial barrier function
87 Barbara,G. et al. (2007) Mast cell-dependent excitation of visceral-nociceptive sensory neurons in
irritable bowel syndrome. Gastroenterology 132, 26-37
88 Alonso,C. et al. (2008) Maladaptive intestinal epithelial responses to life stress may predispose
healthy women to gut mucosal inflammation. Gastroenterology 135, 163-172
89 it-Belgnaoui,A. et al. (2005) Acute stress-induced hypersensitivity to colonic distension depends upon
increase in paracellular permeability: role of myosin light chain kinase. Pain 113, 141-147
90 Demaude,J. et al. (2009) Acute stress increases colonic paracellular permeability in mice through a
mast cell-independent mechanism: involvement of pancreatic trypsin. Life Sci. 84, 847-852
91 Groschwitz,K.R. and Hogan,S.P. (2009) Intestinal barrier function: molecular regulation and disease
pathogenesis. J. Allergy Clin. Immunol. 124, 3-20
92 Geboes,K. and Collins,S. (1998) Structural abnormalities of the nervous system in Crohn's disease and
ulcerative colitis. Neurogastroenterol. Motil. 10, 189-202
93 Neunlist,M. et al. (2003) Changes in chemical coding of myenteric neurones in ulcerative colitis. Gut
52, 84-90
94 Thuijls,G. et al. (2009) Intestinal cytoskeleton degradation precedes tight junction loss following
hemorrhagic shock. Shock 31, 164-169
95 Mittal,A. et al. (2008) The potential role for xanthine oxidase inhibition in major intra-abdominal
surgery. World J. Surg. 32, 288-295
96 Anup,R. et al. (2000) Role of xanthine oxidase in small bowel mucosal dysfunction after surgical
stress. Br. J. Surg. 87, 1094-1101
97 Ramachandran,A. et al. (2001) Intestinal mitochondrial dysfunction in surgical stress. J. Surg. Res. 99,
120-128
98 Lewis,K. and McKay,D.M. (2009) Metabolic stress evokes decreases in epithelial barrier function.
Ann. N. Y. Acad. Sci. 1165, 327-337
99 Holland,J. et al. (2005) Intraoperative splanchnic hypoperfusion, increased intestinal permeability,
down-regulation of monocyte class II major histocompatibility complex expression, exaggerated acute
phase response, and sepsis. Am. J. Surg. 190, 393-400
100 Gatt,M. et al. (2007) Review article: bacterial translocation in the critically ill--evidence and methods
of prevention. Aliment. Pharmacol. Ther. 25, 741-757
101 MacFie,J. et al. (2006) Bacterial translocation studied in 927 patients over 13 years. Br. J. Surg. 93,
87-93
102 MacFie,J. (2004) Current status of bacterial translocation as a cause of surgical sepsis. Br. Med. Bull.
71, 1-11
103 Balzan,S. et al. (2007) Bacterial translocation: overview of mechanisms and clinical impact. J.
Gastroenterol. Hepatol. 22, 464-471
- 165 -
Chapter 6
104 Buchholz,B.M. et al. (2009) Nonhemopoietic cell TLR4 signaling is critical in causing early
lipopolysaccharide-induced ileus. J. Immunol. 183, 6744-6753
105 Derikx,J.P. et al. (2008) New Insight in Loss of Gut Barrier during Major Non-Abdominal Surgery.
PLoS. One. 3, e3954
106 Furlan,R. et al. (2006) Sympathetic overactivity in active ulcerative colitis: effects of clonidine. Am.
J. Physiol Regul. Integr. Comp Physiol 290, R224-R232
107 Martinez,V. and Tache,Y. (2006) CRF1 receptors as a therapeutic target for irritable bowel
syndrome. Curr. Pharm. Des 12, 4071-4088
- 166 -
Chapter
7.
Selective 7 nicotinic
acetylcholine receptor agonists
worsen disease in experimental
colitis
Susanne A. Snoek1, Marleen I. Verstege1, Esmerij P. van der Zanden1, Nigel Deeks2,
David C Bulmer2, Michael Skynner2, Kevin Lee2, Anje A. te Velde1, Guy E.
Boeckxstaens1,3, and Wouter J. de Jonge1
1. Laboratory of Gastroenterology and Hepatology, Academic Medical Centre,
The Netherlands,
2. Immuno-Inflammation CEDD, GlaxoSmithKline Stevenage UK.
3. Catholic University of Leuven, Belgium.
Chapter 7
Abstract
Introduction. In various models vagus nerve activation has been shown to
ameliorate intestinal inflammation, via nicotinic acetylcholine receptors (nAchRs)
expressed on immune cells. As the 7 nAchR has been put forward to mediate this
effect, we studied the effect of nicotine and two selective 7 nAchR agonists (ARR17779,
(-)-spiro[1-azabicyclo[2.2.2]
octane-3,5′-oxazolidin-2′-one
and
GSK1345038A) on disease severity in two mouse models of experimental colitis.
Materials and methods: Colitis was induced by administration of 1.5% dextran
sodium sulphate (DSS) in drinking water or 2 mg 2,4,6-trinitrobenzene sulphonic
acid (TNBS) intrarectally. Nicotine (0.04 and 0.4 mg/kg), AR-R17779 (0.1-5
mg/kg) or GSK1345038A (3-60 mg/kg) was administered daily by i.p. injection.
After 7 (DSS) or 5 (TNBS) days clinical parameters and colonic inflammation were
scored.
Results. Nicotine and both 7 nAchR agonists reduced the activation of NF-B and
pro-inflammatory cytokines in whole blood and macrophage cultures. In DSS
colitis, nicotine treatment reduced colonic cytokine production, but failed to reduce
disease parameters. Reciprocally, treatment with AR-R17779 or GSK1345038A
worsened disease and led to increased colonic pro-inflammatory cytokine levels in
DSS colitis. The highest doses of GSK1345038A (60 mgkg) and AR-R17779 (5
mg/kg) ameliorated clinical parameters, without affecting colonic inflammation.
Neither agonist ameliorated TNBS-induced colitis.
Discussion. Although nicotine reduced cytokine responses in vitro, both selective
7 nAchR agonists worsened the effects of DSS-induced colitis or were ineffective
in those of TNBS-induced colitis. Our data indicate the need for caution in
evaluating 7 nAchR as a drug target in colitis.
- 168 -
Selective 7 nAchR agonists worsen disease in experimental colitis
Introduction
Genetic association studies
2
and functional evidence
3,4
have increased the
recognition that intestinal macrophages play an important role in initiation and
progression of inflammatory bowel disease (IBD). In several studies it was
demonstrated that resident macrophages in mucosal samples of active ulcerative
colitis (UC) and Crohn’s disease (CD) patients phenotypically and functionally
differ from healthy controls
5-7
. Similarly, data obtained from mouse models of
colitis imply a crucial role for macrophages: in IL-10 deficient mice that develop
colitis spontaneously, intestinal inflammation is prevented by the use of antagonists
of chemokine receptors
8
that are generally expressed by macrophages, or by
elimination of tissue macrophages 4. Furthermore, colitis can still be induced in the
absence of T and B cells 3. Recently, it has been shown that macrophage-derived IL10 is crucial for the induction of regulatory T-cells thereby controlling intestinal
inflammation in colitis 9.
Recently, the parasympathetic system, in particular the vagus nerve, has
been shown to negatively regulate macrophage immune responses via the peripheral
10,11
. Activation of the so-called ‘cholinergic anti-
release of acetylcholine
inflammatory pathway’ has been shown to ameliorate disease in various models of
inflammatory disease including, sepsis
and postoperative ileus
11
, ischemia reperfusion
12
, haemorrhage
13
,
14
. In mouse models of IBD and postoperative ileus,
enhanced parasympathetic output is involved in the negative regulation of intestinal
inflammation via efferent activity of the vagus nerve
. Ghia et al. have recently
14-16
demonstrated the eminent role for cholinergic inflammatory control in two
experimental colitis models 15,16. In these studies it was shown that chemical as well
as surgical blockade of vagus nerve signalling significantly worsens colitis and
enhances colonic inflammatory mediators. The anti-inflammatory effect of the vagus
nerve most likely involves activation of the nicotinic acetylcholine receptors
(nAchRs) on immune cells such as macrophages
11,14,17,18,18
or dendritic cells
19,20
.
This notion is supported by clinical observations that smoking, and the
- 169 -
Chapter 7
administration of nicotine (i.e. via patches) may have a protecting effect on colonic
inflammation in UC, even though results are generally disappointing due to the
significant toxic adverse-events 21.
The cellular pathways of nicotinic inhibition of macrophage activation
involve the activation of anti-inflammatory Stat3/Socs3 signalling pathways
14
and
inhibition of NF-B signalling 22. Earlier studies indicate that the anti-inflammatory
effect of acetylcholine is mediated through the α7 nicotinic acetylcholine receptor
(α7 nAchR) 11,18 expressed by human 18,22 and mouse macrophages 18,20,22. Given the
purported role of α7 nAchRs in mediating the cholinergic anti-inflammatory
pathway 14,18,23, selective α7 nAchR agonists may bear more therapeutic potential in
ameliorating disease compared to nicotine. Therefore, we explored the potential of
pharmacological activation of the cholinergic anti-inflammatory pathway by
treatment with nicotine and two α7 nAchR selective agonists in two mouse models
of colitis. In dextran sodium sulphate (DSS) induced colitis, our results show that
nicotine does not affect disease severity. Both selective α7 nAchR agonists ARR17779 and GSK1345038A affect disease severity in a bell-shaped response curve;
low doses aggravate disease, while high doses ameliorate disease. In 2,4,6trinitrobenzene sulphonic acid (TNBS) colitis, treatment with GSK1345038A was
ineffective. These data have important repercussions on the therapeutic potential of
α7 nAchR specific agonists in colitis.
- 170 -
Selective 7 nAchR agonists worsen disease in experimental colitis
Material and Methods
Animals
Female C57BL/6 mice (8-10 weeks old and weighing 20-25 g; Charles River) were
housed and maintained under standard conditions at our animal facility. Food and
water were given ad libitum. All animal experiments were performed according to
the guidelines of the Animal Research Ethics Committee of the University of
Amsterdam.
Induction of colitis
To induce DSS colitis, 1.5% (w/v) DSS (TdB Consultancy, Uppsala, Sweden) was
administered in the drinking water of the mice during 7 days. Body weight was
recorded daily and weight loss as on day 7 as compared to day 0 was calculated.
Animals were killed on day 7 of DSS administration. Hapten-induced colitis was
induced by rectal administration of one dose of 2 mg TNBS (Sigma Chemical Co, St
Louis, MO) in 40% ethanol (Merck, Darmstadt, Germany), using a vinyl catheter
that was positioned 3 cm from the anus. During the instillation, the mice were
anaesthetised
using
isoflurane
(1-chloro-2,2,2-
trifluoroethyl-isoflurane-
difluoromethyl-ether; Abbott Laboratories Ltd., Queenborough, Kent, UK), and
after the instillation mice were kept vertically for 30 seconds. Five days after TNBS
instillation, mice were killed. Mice received a daily intraperitoneal (i.p.) injection
with nicotine (0.04 or 0.4 mg/kg, or 0.25 or 2.5 mol/kg respectively) (SigmaAldrich, Zwijndrecht, the Netherlands); AR-R17779 (0.1, 0.3, 1, 3 or 5 mg/kg; or
0.6-30 mol/kg) (kindly provided by Critical Therapeutics, Lexington, MA) or
GSK1345038 (3, 10, 30 or 60 mg/kg or 6-120 mol/kg) (kindly provided by Glaxo
SmithKline, Stevenage, UK) in 1% methylcellulose. The treatment with agonists
was started at the first day of DSS or TNBS administration.
- 171 -
Chapter 7
GSK1345038A pharmacokinetics
GSK1345038A, 60 or 120 mol/kg, was administered i.p. to C57Bl/6 mice, and
blood samples were taken at time points 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8 and 12 h. Blood
samples were analysed for the free base of GSK1345038A using a method based on
protein precipitation and HPLC-MS/MS analysis. Acetonitrile: ammonium acetate
(10mM) (8:2, 250 µL) containing an appropriate internal standard was added to the
samples of blood (50 µL diluted with 50 µL with water). Samples were mixed
thoroughly (mechanical shaking for 20 minutes), and then centrifuged (2465 x g for
15 minutes at room temperature). An aliquot of the resulting supernatant was
analysed for GSK1345038A by reverse phase HPLC-MS/MS using a heat-assisted
electrospray interface in positive ion mode (Sciex API 4000) and an ACE-3 C18
column (50 x 4.6mm ID, 3m; Hichrom). The mobile phase was delivered as a
linear gradient of 20% to 95% acetonitrile : ammonium acetate (1mM containing
0.1%v/v formic acid) over 0.8 minutes. The final composition was held for 0.8
minutes before return to initial composition. Nominal MRM transition for
GSK1345038A was 454 to 123. Concentration range for the assay was; 0.011 to
22.0 M with a lower limit of quantification (LLQ) of 0.011 µM.
Assessment of colitis
Faecal blood, diarrhoea and disease activity index (DAI) were scored as described in
table 1. The wet weight of each colon was recorded and used as an index of diseaserelated intestinal wall thickening. The total length of the colon was measured and
colon shortening as a consequence of DSS-induced colitis was used as a disease
parameter. Subsequently, the colons were separated from mesentery and fat and
longitudinally divided into two parts for histological examination and measurement
of cytokines.
- 172 -
Selective 7 nAchR agonists worsen disease in experimental colitis
a. weight loss
b. stool consistency
c. bleeding
0: < 1%
0: normal
0: negative
1: 1-5%
2: loose stools
2: positive
2: 5-10%
4: diarrhoea
4: gross bleeding
3: 10-15%
4: >15%
Table 1. Scoring of the Disease Activity Index (DAI). To determine the DAI, scores of a., b. and
c. were combined and divided by three. Bodyweight loss was calculated as the percentage
difference between the body weight on day 0 and the body weight on day 7 of the experiment. The
appearance of diarrhoea is defined as mucus/faecal material adherent to anal fur 1.
Histological examination
The longitudinally divided colons were fixed in 4% formalin and embedded in
paraffin for routine histology. An experienced pathologist microscopically evaluated
formalin-fixed haematoxylin tissue sections in a blinded fashion. Rolled colon was
evaluated, and graded from 0 to 26 points as indicator of incidence and severity of
inflammatory lesions based on the extent of the involved area, the number of follicle
aggregates, oedema, fibrosis, hyperplasia, erosion/ulceration, crypt loss, and
infiltration of granulocytes and mononuclear cells (see table 2).
Score
0
1
2
3
4
Area involved
0%
< 10%
10-25%
25 - 50%
>=50%
Follicles
Normal (0-1)
Little (2-3)
Moderate (4-5)
Extensive (>6)
Oedema
Absent
Little
Moderate
Extensive
Erosion/ulceration
0%
< 10%
10-25%
25 - 50%
Fibrosis
Absent
Little
Moderate
Extensive
Hyperplasia
0%
< 10%
10-25%
25 - 50%
>=50%
Crypt loss
0%
< 10%
10-25%
25 - 50%
>=50%
Granulocytes
Normal
Few
Moderate
Extensive
Mononuclear cells
Normal
Few
Moderate
Extensive
>=50%
Table 2. Histopathology score
- 173 -
Chapter 7
Colonic cytokine production
For cytokine measurements, colons were diluted 1:9 in lysis buffer containing 300
mM NaCl, 30 mM Tris, 2 mM MgCl2, 2 mM CaCl2, 1% Triton X-100, pepstatin A,
leupeptin, and aprotinin (all 20 ng/ml; pH 7.4), and incubated at 4°C for 30 minutes.
Homogenates were centrifuged at 1500 x g at 4°C for 15 minutes, and supernatants
were stored at -20°C until analyses. TNF, IL-6 and IL-17 in supernatants were
analysed by mouse ELISA Duoset kits (R&D Systems, Minneapolis, MN). Assays
were performed according to the manufacturer’s instructions.
Whole blood stimulation assays
Whole blood was taken via heart punction following anaesthesia. Aliquots of 50 l
were divided onto round bottom 96 wells plates and treated with appropriate
concentrations of nicotinic agonists diluted in 50uL of RPMI 1640 supplemented
with 10% heat-inactivated foetal calf serum (FCS; Gibco-BRL, Breda, The
Netherlands), 2 mM L-glutamine, 1000 U/ml penicillin, 1000 g/ml streptomycin,
250 ng/ml amphotericin B (Gibco) for 15 minutes 37oC. Subsequently, heat killed E.
coli (1x104/well) or lipopolysaccharide (LPS) (Sigma) at a final concentration of 100
ng/ml was added to the wells. After 3 hours of stimulation, plates were centrifuged,
supernatants were collected and levels of TNF, IL-6 and IL-17 were analysed by
ELISA (R&D Systems).
NF-B activity assay
Immortalised peritoneal macrophages RAW264.7 were stably transfected with a
NF-B luciferase reporter construct (Clontech, Mountain View, CA) in which a
PDNA3.1(+)-derived neomycin resistance TK cassette was inserted (referred to as
pNF-Bneo-luc). Transfection was performed using Nucleofactor V (Lonza, Cologne,
Germany). Briefly, 0.5 µg per 106 cells of constructs pNF-Bneo-luc was suspended
in 75 µl of 150mM sterile NaCl solution. The transfection was allowed to proceed
- 174 -
Selective 7 nAchR agonists worsen disease in experimental colitis
for 16 hours, and the medium refreshed. Twenty-four hours after transfection,
neomycin resistant clones were selected and subcloned.
For assay, cells were
pretreated with nicotinic agonists at the concentration indicated for 20 minutes,
washed and subsequently stimulated with LPS (100ng/mL; Sigma) for 6 hours. After
treatment, the medium was removed; the cells were washed three times with icecold PBS and lysed with Passive Lysis Buffer supplied in the LuciferaseTM Reporter
Assay Kit (Promega Corporation, Madison, WI) and the lysate was assayed for
luciferase activity according to the manufacturer’s instructions.
Statistics
The values of the groups without DSS and TNBS groups treated with nicotine and
α7 nAchR agonists are relative values (%) as compared to the DSS and TNBS
groups treated with vehicle. Differences between groups were analysed using the
nonparametric Mann-Whitney U test. P<0.05 was considered significant. All
analyses were performed using SPSS (SPSS Inc. Chicago, Ill, USA).
- 175 -
Chapter 7
Results
Macrophage activation is modulated by nicotine, and 7nAchR
agonists GSK1345028A and AR-R17779
Initially, we reproduced experiments showing that nicotine, AR-R17779
14,24
, and
GSK1345028A reduced TNF and IL-6 release in-vitro in Biogel elicited peritoneal
macrophages stimulated with heat-killed E. coli or LPS in a 0-1000nM
concentration range
24
(and data not shown). In line with these previous
observations, AR-R17779 and GSK1345028A, as specific α7 nAchR agonists were
less potent in reducing peritoneal macrophage cytokine release as compared to
nicotine 24 (data not shown). In whole blood cell preparations (see figure 1), nicotine
or the 7 nAchR agonists, AR-R17779 and GSK1345028A, significantly reduced
TNF and IL-6 production in LPS-or heat-killed E. coli activated whole blood cell
preparations, albeit the potency to reduce cytokine production was less pronounced.
None of the three agonists significantly reduced IL-6 production after stimulation
with LPS (see figure 1). The values for these inflammatory substances in
unstimulated cells were below levels of detection (data not shown).
The potential of nicotinic agonists to reduce cytokine production has
previously been associated with inhibition of NF-B activity
23,24
. We explored the
potency of GSK1345028A and AR-R17779 to reduce NF-B transcriptional activity
in activated macrophages. To this end, we investigated the effect of nicotine, ARR17779 and GSK1345028A on NF-B activation induced by LPS in a reporter
assay using the macrophage cell line RAW264.7, which was stably transfected with
a NF-B reporter construct. As shown in figure 2, LPS-induced NF-B
transcriptional activity that was significantly reduced by nicotine, AR-R17779, as
well as GSK1345038A.
- 176 -
Selective 7 nAchR agonists worsen disease in experimental colitis
50
AR-R17779 GSK1345038A
Nicotine
concentration agonist (nM)
125
*
50
Nicotine
*
75
25
Nicotine
AR-R17779 GSK1345038A
* *
50
0
0
100
1000
10000
0
0
100
1000
10000
25
100
concentration agonist (nM)
0
100
1000
10000
* *
E. coli
0
10
100
1000
*
75
TNF (% of control)
100
AR-R17779 GSK1345038A
concentration agonist (nM)
LPS
0
10
100
1000
TNF
TNF (% of control)
125
*
25
0
100
1000
10000
Nicotine
*
50
0
0
100
1000
10000
0
0
100
1000
10000
25
** *
75
0
100
1000
10000
75
E. coli
100
0
100
1000
10000
IL-6 (% of control)
125
0
10
100
1000
LPS
100
0
10
100
1000
IL-6
IL-6 (% of control)
125
AR-R17779 GSK1345038A
concentration agonist (nM)
Figure 1. Cytokine production in mouse whole blood induced by LPS and E. coli. Whole
blood from healthy female C57BL/6 mice was stimulated ex vivo with nicotine (0-1000 nM), ARR17779 (0-10,000 nM), GSK1345038A (0-10,000 nM) and subsequently incubated with LPS (100
ng/ml) (left panels) or E. coli. IL-6 (upper panels) and TNF (lower panels) Values are indicated as
compared to vehicle Data represent three independent experiments. Asterisks indicate significant
differences (*= p<0.05), bars indicate mean ± SEM.
*
75
*
**
**
*
*
*
50
Nicotine (nM)
-LPS
0
1
10
100
1000
10000
0
0
1
10
100
1000
10000
25
EV
NS
0
0.1
1
10
100
1000
NF-kB Activity (%)
100
AR-R17779 (nM) GSK1345038A (nM)
+ LPS
Figure 2. Effects of nicotine, AR-R17779 and GSK1345038A on NF-B activation in
RAW264.7 cells. RAW264.7 cells stably transfected with NF-ĸB luciferase reporter constructs
were pre-treated with different concentrations of nicotine (0-1000 nM), AR-R17779 (0-10,000 nM)
or GSK1345038A (0-10,000 nM) and stimulated with LPS (100 ng/ml). Values are relative as
compared to vehicle. EV= empty vector, NS = not stimulated. Data represent three independent
experiments. Asterisks indicate significant differences (*= p<0.05, **= p<0.01) as compared to
vehicle. Bars represent mean ± SEM.
- 177 -
Chapter 7
Treatment with nicotine does not affect clinical parameters in DSSinduced colitis
Given the reported potential of the vagus nerve to reduce disease in various mice
models, including colitis 15,16, and the positive association of smoking and the course
of UC 25, we next tested whether treatment with nicotine affected the disease course
of DSS–induced colitis. Daily treatment with nicotine did not alter weight loss (see
figure 3a) or DAI (see figure 3b) as compared with the vehicle-treated group. Only
colon weight, which represents thickening of the colon by oedema, was significantly
reduced by treatment with both 0.04 and 0.4 mg/kg nicotine (see figure 3c) but colon
shortening was not affected by nicotine administration (see figure 3d). To test the
effect of nicotine on intestinal inflammation we measured the production of TNF,
IL-6 and IL-17 in colon homogenates. Although TNF levels were not altered,
colonic IL-6 and IL-17 levels were significantly reduced by nicotine treatment (see
figure 3e). However, this reduced cytokine production was not reflected in a
decreased histopathology score (see table 3).
- 178 -
130
175
120
150
125
DAI (%)
110
100
90
100
75
50
25
80
10
0
Nicotine (mg/kg)
-DSS
b.
130
80
110
100
90
80
10
0
Nicotine (mg/kg)
100
*
50
*
*
ve
0.
4
04
4
0.
04
0.
ic
ve
h
0.
4
04
0.
le
hi
c
ve
Nicotine (mg/kg)
*
50
0
le
0
0
100
0.
50
cl
e
100
150
IL-17 (% of vehicle)
150
IL-6 (% of vehicle)
TNF (% of vehicle)
+DSS
-DSS
d.
150
e.
Nicotine (mg/kg)
+DSS
-DSS
0.
04
ve
hi
cl
e
ve
hi
cl
e
0.
4
0.
04
ve
hi
cl
e
70
10
0
120
0.
4
*
**
90
colon length (%)
100
ve
hi
cl
e
colon weight (%)
+DSS
-DSS
110
c.
0.
4
Nicotine (mg/kg)
+DSS
hi
a.
0.
04
ic
le
ic
le
ve
h
0.
4
0.
04
ve
h
ve
h
ic
le
ic
le
0
ve
h
weight (%)
Selective 7 nAchR agonists worsen disease in experimental colitis
Nicotine (mg/kg)
Nicotine (mg/kg)
Figure 3. Effects of nicotine on DSS-induced colitis. a. Percentage body weight on day 7 as
compared with body weight on day 0 of the experiment. b. Disease activity index (DAI) as
described in material and methods. c. Colon length. d. Colon weight per centimetre colon. e. TNF,
IL-6 and IL-17 levels in colon homogenates. Data are expressed as % of mice receiving DSS and
treated with vehicle. Asterisks indicate significant differences (*p<0.05, **p<0.01) as compared to
DSS group treated with vehicle. n= 10 per group. Columns indicate mean ± SEM.
- 179 -
Chapter 7
Nicotine (mg/kg)
vehicle
0.04
0.4
Area involved
2.8 ± 0.30
2.9 ± 0.32
2.8 ± 0.40
Follicles
1.5 ± 0.55
1.5 ± 0.41
0.6 ± 0.22
Oedema
0.6 ± 0.20
0.7 ± 0.20
0.8 ± 0.21
Erosion/ulceration
1.0 ± 0.10
1.3 ± 0.19
1.0 ± 0.18
Crypt loss
2.4 ± 0.33
2.6 ± 0.35
2.5 ± 0.42
Granulocytes
1.4 ± 0.12
1.3 ± 0.19
1.4 ± 0.33
Monocytes
1.8 ± 0.19
1.4 ± 0.12
1.5 ± 0.18
Total score
11.6 ± 1.69
11.7 ± 1.58
10.5 ± 1.94
Table 3. The effect of nicotine on colonic inflammation in DSS-induced colitis. C57BL/6 mice
were administered 1.5% DSS in drinking water and sacrificed at day 7. H&E stainings were
performed on whole colons including rectum from groups treated with vehicle, 0.04 and 0.4 mg/kg
nicotine and scored by an experienced pathologist. H&E stainings of colon and rectum showed
inflammatory features including crypt damage, follicles, oedema, ulceration and influx of
inflammatory cells. Mice per group: n=10. Data represent mean ± SEM.
Treatment with 7nAchR agonists GSK1345038A and AR-R17779
worsened the clinical parameters of colitis
We next questioned whether nicotine treatment failed to reduce disease severity in
DSS-induced colitis because nicotine does not selectively target the α7 nAchR. In
separate experiments, we therefore tested the efficacy of AR-R17779 and
GSK1345038A to ameliorate DSS-induced colitis. A dose of 6 mol/kg of ARR17779 results in a Cmax of 4.6 M and a half life of approximately 150 min (G.R.,
personal communication), and should thus reach the effective concentration range to
reduce cytokine release in macrophages
24
and whole blood (see figures 1 and 2) .
Hence, we administered AR-R17779 at a 0.1-5 mg/kg dose daily. Daily i.p. injection
with 0.3, 1 and 3 mg/kg of AR-R17779 aggravated weight loss (see figure 4a). In
contrast, in the group treated with a highest dose of AR-R17779 (5 mg/kg) weight
loss was prevented (see figure 4a).
- 180 -
130
175
120
150
100
***
90
*
** **
***
*** ***
*
***
**
75
**
25
120
3
10
30
3
60
60
ve
hi
c
ve le
hi
cl
e
0.
1
0.
3
1
3
ve 5
hi
cl
e
3
10
30
60
* *
80
10
0
AR-R17779 (mg/kg) GSK1345038A(mg/kg)
AR-R17779 (mg/kg) GSK1345038A(mg/kg)
+DSS
*
90
ve
hi
c
ve le
hi
cl
e
0.
1
0.
3
70
10
0
100
30
80
110
3
10
90
ve 5
hi
cl
e
colon length (%)
130
110
100
+DSS
3
b.
120
-DSS
ve 5
hi
cl
e
AR-R17779 (mg/kg) GSK1345038A(mg/kg)
-DSS
1
+DSS
1
ve
hi
cl
ve e
hi
cl
e
0.
1
0.
3
60
30
3
10
3
ve 5
hi
cl
e
1
ve
hi
c
v e le
hi
cl
e
0.
1
0.
3
0
-DSS
colon weight (%)
**
**
100
AR-R17779 (mg/kg) GSK1345038A(mg/kg)
c.
***
50
80
10
0
a.
*
125
**
110
DAI (%)
weight (%)
Selective 7 nAchR agonists worsen disease in experimental colitis
d.
-DSS
+DSS
Figure 4. Effects of the α7nAchR agonists AR-R17779 and GSK1345038A on DSS-induced
colitis. a. Percentage body weight on day 7 as compared with body weight on day 0 of the
experiment. b. Disease activity index (DAI) as described in material and methods. c. Colon weight
per centimetre colon. d. Colon length. Data are expressed as % of DSS group treated with vehicle.
Asteriks indicate significant difference (*P<0.05, **P<0.01, ***P<0.001) as compared with DSS
group treated with vehicle. Mice per group: 0.1, 0.3, 1, 3 and 5 mg/kg AR-R17779 and 3, 10 and
30 mg/kg GSK1345038A: n = 10; vehicle groups and 60 mg/kg GSK1345038A: n= 18. Bars
indicate mean ± SEM.
To confirm these data, we next tested another specific α7 nAchR agonist
GSK1345038A in the same model of DSS-induced colitis. Similar to AR-R17779,
we first assessed the optimal dosage range for GSK1345038A by measurement of
the blood concentration of GSK1345038A. The pharmacokinetics indicated that
GSK1345038A has a half live of 2-3 hours, and reaches blood concentrations of 525 µM in a dosage range of 60-120 mol/kg mouse (see figure 5), that is, the
effective dose range to reduce cytokine release in our in vitro assays (see figures 1
- 181 -
Chapter 7
and 2). Hence, to reach optimal circulation levels in vivo, we administered doses of
6, 20, 60, and 120 mol/kg (3, 10, 30 and 60 mg/kg) i.p. daily.
Blood Conc (µM)
25
20
15
10
5
0
0
1
2
3
4
5
6
7
8
9
10
11
12
Time (h)
Figure 5. The time course for the concentrations of GSK1345038A in the blood.
Concentrations of GSK1345038A (60 [closed circles] and 120 [open circles] mol/kg) in mouse
blood was assessed as a function of time. Data shown are the mean ± SEM of triplicate
measurements of 4 mice.
In line with the results obtained using AR-R17779, weight loss was
significantly enhanced by daily injection with 3, 10 or 30 mg/kg GSK1345038A
(see figure 4a). In accord with the effect of the highest dose of AR-R17779 on the
course of colitis, weight loss was prevented by daily treatment with the highest dose
of GSK1345038A (60 mg/kg) tested (see figure 4a).
The effects of both α7nAchR agonists on the DAI paralleled those of the
effects on weight loss as treatment by AR-R17779 significantly enhanced DAI (see
figure 4b). In contrast, DAI was significantly reduced after treatment with the
highest dose of 5 mg/kg AR-R17779 (see figure 4b). Similar results were obtained
by treatment with GSK1345038A in that it significantly worsened disease, as
reflected in DAI, except with the highest dose of 60 mg/kg GSK1345038A, that
ameliorated DAI compared with the vehicle control (see figure 4b). In contrast to
nicotine treatment, the increase of colon weight was unaffected by either of the α7
nAchR agonists (see figure 4c), while the DSS-induced decrease in colon length was
further reduced by AR-R17779 and treatment with GSK1345038A was ineffective
(see figure 4d).
- 182 -
Selective 7 nAchR agonists worsen disease in experimental colitis
The effect of the 7nAchR agonists GSK1345038A and AR-R17779 on
colonic inflammation in DSS-induced colitis
We next measured the effect of α7 nAchR agonists on colonic cytokine production
after 7 days of DSS administration. In line with the augmented disease outcome,
nicotine treatment (see figure 3e), but neither of the 7 nAchR agonists reduced
colonic TNF and IL-6 (see figure 6). In contrast, colonic TNF, IL-6, and IL-17 were
significantly elevated after treatment with AR-R17779, but not GSK1345038A (see
figure 6).
In addition, histopathology scores were assessed for the doses of the α7
nAchR agonists with most pronounced effects on disease severity. As indicated in
table 4, total histopathology scores after treatment with 5 mg/kg AR-R17779 and 60
mg/kg GSK1345038A did not parallel clinical scores as there was no significant
difference between groups. Similar effects were observed by administration of lower
doses of AR-R17779 (0.3 mg/kg) and GSK1345038A (60 mg/kg); clinical outcome
was poorer, but total histopathology scores were not significantly different from
vehicle controls, except for crypt loss that was significantly worsened by treatment
with 0.3 mg/kg AR-R17779 and 60 mg/kg GSK1345038A dosage (see table 4).
- 183 -
Chapter 7
200
TNF (% of vehicle)
*
100
AR-R17779 (mg/kg)
250
150
100
50
60
30
10
100
AR-R17779 (mg/kg)
60
30
le
3
*
60
0
30
3
1
0.
3
0.
1
cl
e
5
ND
0
100
10
*
3
*
100
GSK-1345038A (mg/kg)
200
*
IL-17 (% of vehicle)
200
ve
hi
ve
hi
c
5
3
1
0.
3
cl
e
0.
1
ve
hi
AR-R17779 (mg/kg)
10
0
0
IL-17 (% of vehicle)
GSK-1345038A (mg/kg)
200
*
200
3
ve
hi
cl
e
5
3
1
0.
3
0.
1
0
IL-6 (% of vehicle)
IL-6 (% of vehicle)
ve
hi
cl
e
0
100
ve
hi
cl
e
TNF (% of vehicle)
200
GSK-1345038A (mg/kg)
Figure 6. TNF, IL-6 and IL-17 production in the colon. The effects of treatment with ARR17779 and GSK1345038A at the indicated dose on colonic cytokine production. Asterisks
indicate significant differences (*p<0.05) as compared to vehicle. Agonist treated groups: n = 10;
vehicle groups and 60 mg/kg GSK1345038A: n= 18. Data are expressed as the mean ± SEM.
- 184 -
Selective 7 nAchR agonists worsen disease in experimental colitis
AR-R17779 (mg/kg)
vehicle
0.3
GSK 1345038A (mg/kg)
5
vehicle
30
60
Area involved
2.9 ± 0.10
2.6 ±0.53
2.9 ± 0.1
3.6 ± 0.15
3.8 ± 0.15
3.0 ± 0.24
Follicles
1.1 ± 0.16
1.3 ± 0.45
0.8 ± 0.21
0.8 ± 0.24
0.3 ± 0.17
0.5 ± 0.16
Oedema
1.5 ± 0.33
1.4 ± 0.27
1.3 ± 0.14
1.7 ± 0.16
1.9 ± 0.15
1.4 ± 0.15
Erosion/ulceration
1.8± 0.36
1.4 ± 0.22
1.9 ± 0.11
0.9 ± 0.22
1.3 ± 0.24
1.5 ± 0.21
Crypt loss
2.0 ± 0.15
2.3 ± 0.09*
2.1 ± 0.99
1.6 ± 0.15
2.4 ± 0.24 *
2.2 ± 0.19
Granulocytes
1.1 ± 0.06
0.8 ± 0.13
1.2 ± 0.07
1.6 ± 0.18
1.2 ± 0.17
1.1 ± 0.17
Monocytes
0.7 ± 0.11
0.9 ± 0.10
0.8 ± 0.12
1.3 ± 0.15
1.7 ± 0.14
1.6 ± 0.15
Total score
11.1 ± 0.6
10.7 ± 2.2
11.0 ± 0.3
11.4 ± 1.25
12.6 ± 1.26
11.3 ± 0.8
Table 4: The effect of α7 agonists AR-R17779 and GSK1345038A on colonic inflammation in
DSS-induced colitis. C57BL/6 mice were administered 1.5% DSS in drinking water and sacrificed
at day 7. H&E stainings were performed on whole colons including rectum from groups treated
with vehicle, 0.3 and 5 mg/kg AR-R17779 or 30 and 60 mg/kg GSK1345038A and scored by an
experienced pathologist. H&E stainings of colon and rectum showed inflammatory features
including crypt damage, follicles, oedema, ulceration and influx of inflammatory cells. Mice per
group: Vehicle: n=18; 0.3 mg/kg AR-R17779 and 30 mg/kg GSK1345038A n = 10; 60 mg/kg
GSK1345038A: n = 18. Data represent mean ± SEM.
The effect of the 7 agonist GSK1345038A in a mouse model of acute
TNBS colitis
To investigate whether the effects of α7 nAchR agonists on colitis were specific for
DSS-induced colitis, we tested GSK1345038A in another acute model of colitis,
TNBS-induced colitis. The main read out for this model is colonic inflammation and
not clinical parameters as the mice are allowed to recover after one dose of TNBS.
As indicated in figure 7a, weight loss 5 days after instillation of TNBS was not
significantly different between groups treated with GSK1345038A and vehicle. In
addition, histopathology scores were not significantly altered by treatment with 30
mg/kg and 60 mg/kg GSK 1345038A (see table 5). GSK1345038A treatment did not
alter colonic production of TNF but not of IL-17, while IL-6 production was below
levels of detection (see figure 7b).
- 185 -
Chapter 7
105
weight (%)
GSK1345038A:
100
30 mg/kg
vehicle
95
60 mg/kg
90
85
2
3
Time (days)
100
*
50
150
100
50
60
30
ve
hi
cl
e
60
30
150
100
50
0
0
ve
hi
cl
e
200
60
150
IL-6 below detection
200
30
200
0
b.
250
250
IL-6 (% of vehicle)
TNF (% of vehicle)
250
5
hi
cl
e
a.
4
ve
1
IL-17 (% of vehicle)
0
Figure 7. Effect of 30 and 60 mg/kg 7nAChR agonist GSK1345038A on TNBS-induced
colitis. a. Body weight is shown as percentage of body weight on day 0 of the experiment. b.
Cytokine levels in colon homogenates. Asterisks indicate significant differences (*p<0.05) as
compared to vehicle. Bars indicate mean ± SEM, n=5.
- 186 -
Selective 7 nAchR agonists worsen disease in experimental colitis
GSK 1345038A (mg/kg)
Vehicle
30
60
Area involved
3.7 ± 0.19
3.7 ± 0.18
2.9 ± 0.26
Follicles
1.0 ± 0.24
1.1 ± 0.40
0.4 ± 0.20
Oedema
0
0.2 ± 0.14
0.3 ± 0.10
Erosion/ulceration
0.4 ± 0.19
0.6 ± 0.32
0.7 ± 0.36
fibrosis
0.7 ± 0.20
0.6 ± 0.30
0.7 ± 0.21
Hyperplasia
3.7 ± 0.20
2.6 ± 0.69
3.1 ± 0.34
Crypt loss
0.8 ± 0.15
1.4 ± 0.48
0.5 ± 0.20
0
0.6 ± 0.17 *
0.3 ± 0.14
Monocytes
0.8 ± 0.15
1.1 ± 0.13
0.9 ± 0.20
Total score
11.1 ± 1.3
11.9 ± 2.8
9.8 ± 2.0
Granulocytes
Table 5. The effect of 7 agonists AR-R17779 and GSK1345038A on colonic inflammation in
TNBS-induced colitis. Mice (n = 7 per group) received one dose of 2 mg TNBS in 30% ethanol
intrarectally and were killed after 5 days. Vehicle, 30 or 60 mg/kg GSK1345038A was injected
daily. H&E stainings were performed on whole colons including rectum and scored by an
experienced pathologist. H&E stainings of colon and rectum showed inflammatory features
including crypt damage, follicles, oedema, ulceration, hyperplasia and influx of inflammatory cells.
Asterisks indicate significant differences (*P < 0.05) as compared with vehicle. Data represent
mean ± SEM.
- 187 -
Chapter 7
Discussion
IBD patients suffer from chronic and relapsing intestinal inflammation, initiated by
aberrant responses of the innate immune system 26,27. Recently, a number of animal
studies demonstrate that innate immune responses are attenuated by stimulation of
the efferent arm of the vagus nerve through its neurotransmitter acetylcholine that
acts on nAchRs, in particular the α7 nAchR, on resident macrophages 11,18,28,28,29.
In various mouse models of inflammatory disease, we
18,22
14,24,30
and others
, observed anti-inflammatory effects of vagus nerve stimulation, as well as
pharmacological stimulation of the cholinergic system by administration of nicotine
and α7 nAchR agonists. In the current study, we aimed to extend these studies by
treating experimental colitis through targeting α7 nAchRs with nicotine, and two
selective α7 nAchR agonists AR-R17779 and GSK1345038A. The agonists were
tested in two mouse models of acute colitis induced by DSS or TNBS. In vitro,
nicotine reduces macrophage NF-B activity and cytokine release significantly. In
addition, treatment of DSS-induced colitis with nicotine led to a significant
reduction in colonic oedema and colonic IL-6 and IL-17 production. However, this
reduction was not marked enough to be reflected in clinical parameters and
histopathological scores. The histopathological scores are the end point of the
inflammatory reaction and contribute greatly to the functionality of the colon and
thus have a large influence on clinical outcome. In addition, reduced IL-17 levels do
not strictly imply reduced disease activity and, recently, data have been obtained
indicating that IL-17 might act as an anti-inflammatory cytokine in the gut 31.
UC Patients with a history of smoking usually acquire their disease after
they have stopped smoking 32-34. Patients who smoke intermittently often experience
improvement in their colitis symptoms during the periods when they smoke
25,33,35
.
Following this reasoning and given the previous reports on the positive effect of
cholinergic activation in experimental models of DSS colitis 15,16, nicotine treatment
may well be beneficial in UC. Indeed, in patient studies treatment with transdermal
- 188 -
Selective 7 nAchR agonists worsen disease in experimental colitis
nicotine was effective for the induction of disease remission in UC patients 21, but
the number of patients that suffered from adverse effects was significantly higher in
the nicotine-treated patient groups 21 as compared with patients treated with standard
therapy. It should be noted that smoking in CD patients worsens the disease.
In the current study, besides nicotine, we tested two selective α7 nAchR
receptor agonists, AR-R17779
36
and GSK1345038A, in the mouse model of DSS
and induced colitis. Treatment with the α7 nAchR agonists both displayed a bellshaped dose-response curve; the highest doses of AR-R17779 and GSK1345038A
significantly ameliorated clinical parameters, whereas lower doses of both
compounds worsened or did not affect clinical parameters. Although our data
confirm the capacity of AR-R17779 and GSK1345038A to reduce pro-inflammatory
mediator release in vitro in macrophage cultures 24 and whole blood, the reduction in
cytokines by nicotine as well as both 7 agonists was around 20–40%, which proved
not to affect disease outcome in the colitis models used in this study.
However, it is possible that at the highest dose the 7 nAchR agonists
might exhibit off-target activity and lose their selectivity for the 7 nAchR, thereby
affecting disease in an 7 nAchR-independent manner. This is also indicated by the
marked dose–response relationship observed between colonic IL-17 levels and ARR17779 treatment, which may be explained by concentration-dependent off-target
activity of AR-R17779. It should be borne in mind that nAchRs are expressed
peripherally as well as centrally and that activation of nAchR on neurones can have
analgesic effects, or modify mucus production, gut motility and blood flow to the
gut
25,37
. In the DSS colitis model, these effects might control food intake and
formation of stools thereby influencing disease activity parameters independently of
the severity of colonic inflammation. Another effect of nAchR activation can be a
change in muscle tone thereby reducing colon length. This might play a role in the
significant reduction of colon length we observed on treatment with AR-R17779.
- 189 -
Chapter 7
In addition, activation of nAchRs plays a role in regulating epithelial
permeability
37,38
and bacterial clearance
39,40
, important factors in the development
of colitis that were not assessed in our experiments. Thus, nAchR activation can
have a variety of effects on disease, independent of immune mediation, because of
its widespread expression on different cell types as well as on different tissue types.
Although we report here that treatment with nicotine, or selective 7 nAchR
agonists, is not effective in experimental colitis, enhanced vagus nerve output has
been shown to reduce inflammation in various mouse models
11,24,28,29,40,41
. This
cholinergic anti-inflammatory effect seems to rely on the expression of the 7
nAchR on innate immune cells 11,18. Reciprocally, in mouse models of colitis, it has
been shown that vagotomy worsens colitis, an effect that was shown to be
counteracted by nicotine administration
15,15,16,42
. Of note in the interpretation of
these studies is that the vagus nerve only marginally innervates the distal colon,
making direct effects of Ach on colonic immune cells unlikely. The vagus nerve
probably relays its immune modulatory effects to the colon in an indirect fashion,
that is, via postganglionic activation or by targeting alternative cell types. Of interest
in this respect is a more recent study in which vagotomy was shown to worsen DSS
colitis due to an impaired potential of antigen presenting cells to induce regulatory
T-cells 42. Notably, the physiological effects of vagus nerve stimulation or vagotomy
as compared with pharmacological activation of acetylcholine receptors differ
greatly, especially when taking into account the changes in sympathetic output. In
addition, vagus nerve stimulation or vagotomy will not only target nAchRs, but also
influence the release of a number of neurotransmitters in the gut that regulate
immune functions, and gut functions such as permeability and blood flow that
possibly influence disease outcome.
Irrespectively, however, nicotine administration ameliorated disease in
previous studies of experimental colitis
15,43
. We cannot explain why the
effectiveness of nicotine to reduce disease parameters was less pronounced in our
study. The nicotine dose used in this study had been shown to be effective at
reducing inflammation in other models of inflammation
- 190 -
24,44
, but possibly, in our
Selective 7 nAchR agonists worsen disease in experimental colitis
colitis experiments a higher dose is required. However, in mice, a higher dose would
result in adverse effects because of activation of a broad range of receptors both
peripherally and centrally
expressed
46
45
. In addition, a large array of nAchRs subtypes are
, and previous studies point towards a role in modulation of intestinal
inflammation for nAchRs containing 5
47
or 42
20
. Thus, the nAchR subtype
involved in the immunomodulatory properties of the vagus nerve remains to be
established.
Alternatively, the outcome of animal experiments with nAchR agonists
could be dependent on the model of inflammation studied, as expression of the
nAchR might vary depending on tissues and cell types involved in disease
development. Notably, in other studies, nicotine treatment worsened the course of
jenunitis in rodent models
among colitis models
47
and TNBS colitis
48
. There are notable differences
49
, which might be important in the effectiveness of the
administered agents. Thus, the effects of nicotine and 7 nAchR agonists may
depend on many experimental factors such as the dose used, administration method,
disease severity and disease model. We conclude that in developing a strategy for
treating colitis using cholinoceptor agonists we should keep in mind that the
expression of nAchRs is extremely widespread both centrally and peripherally. In
addition, the expression of the various nAchRs subtypes on a particular target cell
should be carefully investigated before evaluating the effectiveness of 7 nAchRs as
a drug target in colitis patients.
- 191 -
Chapter 7
Acknowledgements:
The authors gratefully acknowledge grant support from Top Institute Pharma (T1215), and the 6th European Community Framework Program Marie Curie (WdJ).
GEB (VICI), MIV (VICI written by GEB) and WdJ (VIDI) were supported by
grants from the Netherlands Organisation for Scientific Research (NWO), and GEB
by a grant (Odysseus program, G.0905.07) of the Flemish “Fonds Wetenschappelijk
Onderzoek” (FWO). Gregory LaRosa (Bikam Pharmaceuticals, Inc, Fl) is gratefully
acknowledged for supplying the AR-R17779 compound.
- 192 -
Selective 7 nAchR agonists worsen disease in experimental colitis
References
1 Cooper,H.S. et al. (1993) Clinicopathologic study of dextran sulfate sodium experimental murine
colitis. Lab Invest 69, 238-249
2 Barrett,J.C. et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for
Crohn's disease. Nat. Genet. 40, 955-962
3 Dieleman,L.A. et al. (1994) Dextran sulfate sodium-induced colitis occurs in severe combined
immunodeficient mice. Gastroenterology 107, 1643-1652
4 Watanabe,N. et al. (2003) Elimination of local macrophages in intestine prevents chronic colitis in
interleukin-10-deficient mice. Dig. Dis. Sci. 48, 408-414
5 MacDermott,R.P. (1996) Alterations of the mucosal immune system in inflammatory bowel disease. J.
Gastroenterol. 31, 907-916
6 Rogler,G. et al. (1999) T-cell co-stimulatory molecules are upregulated on intestinal macrophages from
inflammatory bowel disease mucosa. Eur. J. Gastroenterol. Hepatol. 11, 1105-1111
7 Kamada,N. et al. (2005) Abnormally differentiated subsets of intestinal macrophage play a key role in
Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J.
Immunol. 175, 6900-6908
8 Tokuyama,H. et al. (2005) The simultaneous blockade of chemokine receptors CCR2, CCR5 and
CXCR3 by a non-peptide chemokine receptor antagonist protects mice from dextran sodium sulfatemediated colitis. Int. Immunol. 17, 1023-1034
9 Murai,M. et al. (2009) Interleukin 10 acts on regulatory T cells to maintain expression of the
transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178-1184
10 Tracey,K.J. et al. (2001) Mind over immunity. FASEB J. 15, 1575-1576
11 Borovikova,L.V. et al. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response
to endotoxin. Nature 405, 458-462
12 Sadis,C. et al. (2007) Nicotine protects kidney from renal ischemia/reperfusion injury through the
cholinergic anti-inflammatory pathway. PLoS. ONE. 2, e469
13 Luyer,M.D. et al. (2005) Nutritional stimulation of cholecystokinin receptors inhibits inflammation via
the vagus nerve. J. Exp. Med. 202, 1023-1029
14 de Jonge,W.J. et al. (2005) Stimulation of the vagus nerve attenuates macrophage activation by
activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 6, 844-851
15 Ghia,J.E. et al. (2006) The vagus nerve: a tonic inhibitory influence associated with inflammatory
bowel disease in a murine model. Gastroenterology 131, 1122-1130
16 Ghia,J.E. et al. (2007) The protective effect of the vagus nerve in a murine model of chronic relapsing
colitis. Am. J. Physiol Gastrointest. Liver Physiol 293, G711-G718
- 193 -
Chapter 7
17 Matsunaga,K. et al. (2001) Involvement of nicotinic acetylcholine receptors in suppression of
antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila
infection by nicotine. J. Immunol. 167, 6518-6524
18 Wang,H. et al. (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of
inflammation. Nature 421, 384-388
19 Nouri-Shirazi,M. et al. (2007) Nicotine alters the biological activities of developing mouse bone
marrow-derived dendritic cells (DCs). Immunol. Lett. 109, 155-164
20 Kawashima,K. et al. (2007) Expression and function of genes encoding cholinergic components in
murine immune cells. Life Sci. 80, 2314-2319
21 McGrath,J. et al. (2004) Transdermal nicotine for induction of remission in ulcerative colitis.
Cochrane. Database. Syst. Rev., CD004722
22 Wang,H. et al. (2004) Cholinergic agonists inhibit HMGB1 release and improve survival in
experimental sepsis. Nat. Med. 10, 1216-1221
23 Ulloa,L. (2005) The vagus nerve and the nicotinic anti-inflammatory pathway. Nat. Rev. Drug Discov.
4, 673-684
24 The,F.O. et al. (2007) Activation of the cholinergic anti-inflammatory pathway ameliorates
postoperative ileus in mice. Gastroenterology 133, 1219-1228
25 Birrenbach,T. and Bocker,U. (2004) Inflammatory bowel disease and smoking: a review of
epidemiology, pathophysiology, and therapeutic implications. Inflamm. Bowel. Dis. 10, 848-859
26 Bouma,G. and Strober,W. (2003) The immunological and genetic basis of inflammatory bowel
disease. Nat. Rev. Immunol. 3, 521-533
27 Xavier,R.J. and Podolsky,D.K. (2007) Unravelling the pathogenesis of inflammatory bowel disease.
Nature 448, 427-434
28 de Jonge,W.J. and Ulloa,L. (2007) The alpha7 nicotinic acetylcholine receptor as a pharmacological
target for inflammation. Br. J. Pharmacol. 151, 915-929
29 van der Zanden,E.P. et al. (2009) The vagus nerve as a modulator of intestinal inflammation.
Neurogastroenterol. Motil. 21, 6-17
30 van Maanen,M.A. et al. (2009) Stimulation of nicotinic acetylcholine receptors attenuates collageninduced arthritis in mice. Arthritis Rheum. 60, 114-122
31 Ogawa,A. et al. (2004) Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced
colitis in mice. Clin. Immunol. 110, 55-62
32 Ingram,J.R. et al. (2004) Nicotine enemas for treatment of ulcerative colitis: a study of the
pharmacokinetics and adverse events associated with three doses of nicotine. Aliment. Pharmacol. Ther.
20, 859-865
33 Pullan,R.D. et al. (1994) Transdermal nicotine for active ulcerative colitis. N. Engl. J. Med. 330, 811815
- 194 -
Selective 7 nAchR agonists worsen disease in experimental colitis
34 Thomas,G.A. et al. (1995) Transdermal nicotine as maintenance therapy for ulcerative colitis. N. Engl.
J. Med. 332, 988-992
35 Sandborn,W.J. (1999) Nicotine therapy for ulcerative colitis: a review of rationale, mechanisms,
pharmacology, and clinical results. Am. J. Gastroenterol. 94, 1161-1171
36 Mullen,G. et al. (2000) (-)-Spiro[1-azabicyclo[2.2.2]octane-3,5'-oxazolidin-2'-one], a conformationally
restricted analogue of acetylcholine, is a highly selective full agonist at the alpha 7 nicotinic acetylcholine
receptor. J. Med. Chem. 43, 4045-4050
37 Thomas,G.A. et al. (2005) Mechanisms of disease: nicotine--a review of its actions in the context of
gastrointestinal disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2, 536-544
38 McGilligan,V.E. et al. (2007) Hypothesis about mechanisms through which nicotine might exert its
effect on the interdependence of inflammation and gut barrier function in ulcerative colitis. Inflamm.
Bowel. Dis. 13, 108-115
39 Zanden,E.P. et al. (2009) Vagus nerve activity augments intestinal macrophage phagocytosis via
nicotinic acetylcholine receptor alpha4beta2. Gastroenterology
40 van Westerloo,D.J. et al. (2005) The cholinergic anti-inflammatory pathway regulates the host
response during septic peritonitis. J. Infect. Dis. 191, 2138-2148
41 Luyer,M.D. et al. (2004) High-fat enteral nutrition reduces endotoxin, tumor necrosis factor-alpha and
gut permeability in bile duct-ligated rats subjected to hemorrhagic shock. J. Hepatol. 41, 377-383
42 O'Mahony,C. et al. (2009) Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive
transfer. Am. J. Physiol Regul. Integr. Comp Physiol 297, R1118-R1126
43 Sykes,A.P. et al. (2000) An investigation into the effect and mechanisms of action of nicotine in
inflammatory bowel disease. Inflamm. Res. 49, 311-319
44 van Maanen,M.A. et al. (2009) The cholinergic anti-inflammatory pathway: towards innovative
treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 229-232
45 Matta,S.G. et al. (2007) Guidelines on nicotine dose selection for in vivo research.
Psychopharmacology (Berl) 190, 269-319
46 Wessler,I. and Kirkpatrick,C.J. (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic
system in humans. Br. J. Pharmacol. 154, 1558-1571
47 Eliakim,R. et al. (2002) Chronic nicotine administration differentially alters jejunal and colonic
inflammation in interleukin-10 deficient mice. Eur. J. Gastroenterol. Hepatol. 14, 607-614
48 Eliakim,R. et al. (1998) Effect of chronic nicotine administration on trinitrobenzene sulphonic acidinduced colitis. Eur. J. Gastroenterol. Hepatol. 10, 1013-1019
49 te Velde,A.A. et al. (2006) Critical appraisal of the current practice in murine TNBS-induced colitis.
Inflamm. Bowel. Dis. 12, 995-999
- 195 -
Chapter 7
- 196 -
Chapter
8.
Acetylcholine protects against
inflammatory cytokine induced
epithelial barrier dysfunction
Marleen I. Verstege1, Shobhit Dhawan1, Linette E. Willemsen2, Jogchum Plat3,
Rene M. van den Wijngaard1, Guy E. Boeckxstaens1,4, Wouter J. de Jonge1
1. Tytgat Institute for Liver and Intestinal Research, Academic Medical
Centre, Amsterdam, The Netherlands
2. Department of Pharmacology and Pathophysiology, Utrecht Institute for
Pharmaceutical Sciences, Faculty of Science, Utrecht University, The
Netherlands
3. Department of Human Biology, Nutrition and Toxicology Research Institute
Maastricht (NUTRIM), Maastricht University, Maastricht, The Netherlands
4. Department of Gastroenterology, Catholic University of Leuven, Leuven,
Belgium
Chapter 8
Abstract
Background: Increased epithelial permeability during chronic inflammation is a
perpetuating factor in inflammatory bowel disease. Pro-inflammatory cytokines
(TNF- and IL-1) are shown to increase epithelial permeability. In animal models,
vagus nerve-derived acetylcholine (Ach) ameliorates intestinal inflammation via
activation of cholinergic receptors on innate inflammatory cells.
Aim: To investigate whether Ach confers protection to intestinal inflammation via
modulation of chemokine release and maintenance of barrier function in human
epithelial cells.
Methods: Monolayers of Caco-2 cells were incubated with cholinergic agonists
and/or an inflammatory cytokine mix (TNF-/IL-1) and changes in IL-8
production, transepithelial electrical resistance (TEER), and transepithelial FITCdextran flux were analysed in co-culture systems. Occludin and ZO-1 expression
were analysed by immunoblot and immunofluorescence, respectively.
Results: Ach and muscarine, but not nicotine reduced IL-1-induced NF-B activity
in Caco-2 cells. This effect was mediated via mAChRs, as muscarine and Ach were
equally potent in reducing IL-8 while nicotine was not effective. In conjunction,
IL1-or TNF- induced IL-8 production by Caco-2 cells was inhibited by Ach in a
dose-dependent fashion. Ach enhanced epithelial permeability under steady state
conditions. On the other hand, Caco-2 cells exposed to cytokines (IL-1/TNF-) for
72 hours showed a reduced barrier function reflected in a reduced TEER, increased
dextran flux and loss of ZO-1 expression, and this impaired barrier integrity was
counteracted by Ach and muscarine, but not nicotine.
Conclusion: Ach protects epithelial cells from the detrimental effects of IL-1 and
TNF- on the integrity of the intestinal epithelial barrier via activation of mAChRs.
- 198 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
Introduction
The intestinal epithelial layer has developed complex barrier mechanisms to prevent
unrestricted access of luminal contents in the lamina propria, while still allowing
dendritic cell-mediated surveillance of luminal antigens. Barrier proteins involved in
this process include adherens junctions, desmosomes, gap junctions and tight
junctions (TJs). TJs or zonula occludens are the most apical components of these
intercellular junctions. The main functions of the TJs are to prevent diffusion of
membrane proteins and lipids between basolateral and apical membranes so that cell
polarity is preserved (fence function) and to function as a selective barrier to
paracellular transport (barrier function). It has been demonstrated that intestinal
epithelial barrier function is impaired in a range of inflammatory disease states such
as inflammatory bowel diseases (IBD), irritable bowel syndrome, post-operative
ileus, celiac disease, food allergy, asthma and rheumatoid arthritis
1-4
. The impaired
epithelial barrier function is likely to contribute to the severity of chronic
inflammation.
TJs are composed of a complex of membrane proteins, including occludin,
several members of the claudin family and junctional adhesion molecule-1 5-12 and a
family of zonula occludens (ZO) proteins that connect the membrane part to the
actin filaments. It has been shown that pro-inflammatory cytokines, such as tumour
necrosis factor (TNF)-, interleukin (IL)-1, interferon (IFN)- and IL-8 increase
intestinal permeability by rearrangement and internalisation of TJ proteins, probably
mediated by a rapid activation of transcription factor NF-B
13-19
. Bacterial
recognition occurs on enterocytes and colonocytes via their apical expression of a
range of Toll-like receptors (TLRs), but epithelial cells generally show enhanced
barrier integrity on TLR ligation, via enhanced TJ expression and rearrangement 20,21.
Intestinal barrier function is highly regulated by neuronal factors, such as
enteric glial cells (EGCs) 22. In addition, recently it has been shown that enhanced
vagus nerve motor output, either stimulated pharmacologically, electrically, or via
- 199 -
Chapter 8
high fat nutrition 23, serves a protective effect on barrier function under conditions of
intestinal inflammation. Cholinergic activity however acts as a double-edged sword:
although protective in inflamed conditions, under healthy conditions vagus nerve
efferent activation was shown to enhance bacterial translocation and paracellular
permeability in the gut 24-26 and Ach increases transcellular transport via muscarinic
Ach receptor (mAchR) activation
27
. These data seem contradictory since an
increased intestinal epithelial barrier leads to an increased exposure of antigens to
the mucosal compartment.
Thus, vagal- or cholinergic activity may favour an enhanced immune
surveillance under healthy conditions, while maintaining barrier function in disease.
Hence we investigated molecular mechanisms of functional changes of cholinergic
modulation of the barrier function and focussed on healthy and inflamed conditions.
In the human intestinal epithelial cell line Caco-2 we demonstrate that cholinergic
agonists reduce TNF- or IL1- induced NF-B activation and IL-8 production.
This effect is mediated via mAchR, since only Ach and muscarine decreases TNF-
and IL-1-induced IL-8 production. Nicotine, which acts on nicotinic acetylcholine
receptors (nAchRs), decreases only IL-8 production that is induced by low
concentrations of IL-1. Moreover, while Ach and muscarine, reduce IL-1-induced
NF-B activation, nicotine fails to counteract NF-B activity. In cytokine stimulated
epithelial cells, Ach protects against enhanced transcellular transport and distorted
ZO-1 expression.
- 200 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
Materials and methods
Cell culture
Caco-2 cells (kindly provided by J. Plat 28, University of Maastricht, Maastricht, The
Netherlands) were cultured in DMEM, 10% foetal calf’s serum (heat inactivated),
5% glutamine and 5% penicilline/streptavidine (all obtained from Gibco BRL,
Breda, The Netherlands) in a 75 cm2 flask. Adherent cultures passased weekly at
subconfluence after trypsinisation. The cells were maintained at 37°C in an
atmosphere of 5% CO2.
After culturing Caco-2 cells in a flask, they were cultured in 12 or 24-wells
plates or transwells (Costar, Cambridge, MA, USA). Cells were used for further
experiments between 14 and 21 days of confluence or when transepithelial electrical
resistance (TEER) was constant over time. The TEER in transwells was measured
with a Millipore-ERS metre (Millipore corp., Bedford, MA).
NF-B reporter assay
Caco-2 cells that were stably transfected with an NF-B luciferase reporter construct
were cultured in 12-wells plate and the cells were washed with PBS followed by
refreshing the medium approximately 2 to 3 hours before the experiment. At the
start of the experiment, the medium was replaced by medium containing 25ng/ml
IL-1 and 10, 100 or 1000 nM Ach (Sigma, St. Louis., MO). One well was kept
blank to serve as a treatment control. Thereafter the cells were incubated at 37°C for
16 hours. Cells were washed with PBS and passive lysis buffer (Promega, Madison,
WI) was added to the cells followed by scraping the attached cells from the dish.
The cell lysates were stored at -80°C until further use.
Luciferase Assay Reagent (Promega) was dispensed into a luminometer
plate well (Grenier Bio One ltd. Stonehouse, UK). The luminometer was
programmed to perform a 2-second measurement delay followed by a 10-second
- 201 -
Chapter 8
measurement read for luciferase activity (sensitivity 240). Cell lysate was added to
the Luciferase Assay Reagent in the luminometer plate and was placed in the
luminometer to measure luciferase activity.
IL-8 measurement and isolation of protein
Caco-2 cells were cultured in 24-wells plates and between 14 and 21 days after
confluence, the cells were stimulated with 0, 100, 1000 or 10.000 nM Ach, nicotine
or muscarine (all from Sigma) for 20 minutes. Thereafter, Ach, nicotine or
muscarine were washed away with PBS and the medium was replaced by DMEM
containing different concentrations of IL-1 (Miltenyi), TNF- (Miltenyi) or no
stimuli and in case of Ach stimulation 50nM of neostigmine (Sigma) may be added
to protect intrinsic Ach from breakdown. After 3 hours, supernatant was collected to
perform an IL-8 ELISA (Arcus Biologicals Srl, Modena, Italy) according to
manufacturer’s manual.
Furthermore, membrane and cytosolic fractions were separated to perform
western blots. Therefore, Caco-2 cells were scraped in PBS (4°C) and centrifuged at
1000 rpm for 5 minutes. Supernatant was removed and lysisbuffer was added to the
cell pellet. The lysisbuffer was made of in 2mM EDTA (Sigma), 250mM mannitol
(Sigma), 20mM Hepes/Tris pH 7.4 (Sigma) and protease inhibitors (Gibco). The cell
pellet was left in lysisbuffer for 30 minutes on ice followed by douncing 30 times.
This homogenate was centrifuged at 1000 rpm for 5 minutes. Supernatant was
collected in an ultracentrifuge tube and centrifuged in an ultracentrifuge at 45000
rpm for 45 minutes. This collected supernatant contains the cytosolic fraction,
whereas the pellet contains the membrane fraction. Membrane fractions were solved
in PBS and to solve the fractions, a 21-gauge needle was used. All the protein
fractions were stored at -20°C.
- 202 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
FITC-dextran permeability determination
Caco-2 cells were cultured on transwells for 72 hours in the presence or absence of a
cytokine cocktail containing 50 ng/ml TNF-, 10 ng/ml IL-1 and 50 ng/ml IFN-.
Simultaneously, the cells were incubated with 0, 100 or 1000 nM Ach with or
without 50nM neostigmine. One hour prior to flux experiment the media of all wells
was refreshed with conditioned medium without phenol red (Gibco). At the mucosal
compartment, 5l FITC-dextran stock (25 mM in MEM- Sigma) was added. At
t=30 and t=60 sample volumes of 100 l were taken from the serosal compartment
and collected in a white 96 wells plate (Corning, Lowell, MA) covered with
aluminium foil. The samples were measured with the Fluoroskan Ascent FL
(Thermo Scientific, Rockford, IL) (ex 492 em 518).
SDS-PAGE and western blotting
Protein contents of membrane and cytosol fractions were determined by the BCA
assay (Pierce, Rockford, IL) according to the manufacturer’s recommendation.
Equal amounts of protein of membrane and cytosol extracts were separated on a
10% sodium dodecyl sulphate-polyacrylamide gel (SDS-PAGE) and electroblotted
to a polyvinylidene difluoride (PVDF) membrane (Millipore). The membrane was
blocked in 5% (w/v) non-fat dry milk (Fluka, St. Louis., MO) in TBS. Thereafter,
the membrane was incubated with primary antibody against occludin (1:1000, rabbit
anti occludin, Zymed Laboratories Inc., South San Francisco, California, USA),
which was dissolved in 1% non-fat dry milk in TBST, at 4°C overnight. After
washing with TBS, the membrane was incubated with horseradish peroxidaseconjugated anti-rabbit Ig antibody (Dako, Glostrup, Danmark) dissolved in 1% milk
in TBST at room temperature for 1 hour. Finally, lumi-light substrate (Roche, Basel,
Switzerland) was used to visualise the western blots. Pictures were made with the
Lumi-Imager F1 (Roche). Equal protein loading was confirmed by western blots for
-actin.
- 203 -
Chapter 8
Immunofluorescence
Caco-2 cells grown in transwells were fixed in 4% paraformaldehyde for 10 minutes.
After pre-incubation with PBS, samples were blocked in 1% bovine serum albumine
(BSA) (Sigma) and 0.1% triton (Sigma) in PBS for 15 minutes. Thereafter, the
samples were incubated with polyclonal antibodies against ZO-1 (1:500, rabbit antiZO-1, Zymed Laboratories Inc., South San Francisco, California, USA) in 1% BSA
and 0.1% triton in PBS for one hour at room temperature. After washing the samples
with PBS, the samples were incubated with the secondary antibody (1:1000, goat
anti-rabbit Alexa 546) in 1% BSA and 0.1% triton in PBS for 30 minutes. Finally,
the samples were washed with PBS and covered with moviol, including DAPI.
Immunostaining and imaging. Confocal immune fluorescence on intestinal
sections was performed as described earlier. The polyclonal antibody against
LAMP-2 was kindly provided by S. Heinsbroek, University of Amsterdam.
Electrical vagal nerve stimulation and in vivo uptake
Electrical vagal nerve stimulation (VNS): Mice were anaesthetised by i.p. injection
of a mixture of Fentanyl Citrate / Fluanisone (Hypnorm; Janssen, Beerse, Belgium)
and Midazolam (Dormicum; Roche, Mijdrecht, The Netherlands). VNS was
performed as described previously 29. In short: the right cervical vagal branch was
prepared free from the carotid artery and ligated with 6-0 silk suture. The part distal
from the ligation was attached to an electrode and 5V stimuli with a frequency of
5Hz, duration of 2ms10 were applied for 5 minutes. In sham mice the cervical skin
was opened and left for 30 minutes covered by moist gauze. After a 30 minutes
recovery period, the in vivo uptake assay was initiated as described below.
In vivo uptake: Surgical procedures were performed under sterile conditions.
Mice underwent a laparotomy and an ileum segment 3-10 cm proximal from the
caecum was opened and its lumen rinsed with pre-warmed (37°C) oxygenated Krebs
buffer. The ileum segment was filled with 1-2 ml of buffer containing FITC-labelled
- 204 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
E.faecium bacteria and clamped at both sides. After 30 minutes the clamped
intestinal
segment
was
removed,
washed
in
PBS
and
processed
for
immunohistochemistry.
Statistical analysis
All data are expressed as the means ± the standard error of the mean (SEM) and
were analysed using Graphpad prism 4 (Graphpad Prism v. 4 for Windows,
GraphPad Software, San Diego, California USA). Differences between groups were
analysed using the non-parametric Mann Whitney U test. All statistics were
performed two-tailed and values of p<0.05 were considered statistically significant
(* p<0.05; ** p<0.01, *** p<0.001).
- 205 -
Chapter 8
Results
Acetylcholine and muscarine inhibit NF-B activity in Caco-2 cells
Since it has been shown that Ach inhibits the NF-B pathway in immune cells 30-36,
we wanted to investigate whether Ach is also able to inhibit NF-B activity in
intestinal epithelial cells. Therefore, an NF-B reporter assay was performed to test
whether Ach, nicotine and muscarine inhibit NF-B activity of Caco-2 cells induced
by IL-1. It seems that Ach and muscarine inhibit NF-B activity in a dosedependent manner (see figure 1). This effect is less pronounced if Caco-2 cells are
incubated with nicotine. These results indicate that mainly the mAchRs are involved
in the inhibition of the NF-B pathway in Caco-2 cells.
Luciferase activity
6.0×10 5
Ach
Muscarine
Nicotine
5.0×10 5
4.0×10 5
0
10
10
10 0
00
0
10
10
10 0
00
0
10
10
10 0
00
3.0×10 5
Concentration (nM)
Figure 1. NF-B reporter assay. Caco-2 cells stably transfected with an NF-B luciferase reporter
construct were incubated with 25ng/ml IL-1 and 10, 100 or 1000 nM Ach, nicotine or muscarine
for 16 hours. Thereafter, luciferase activity was measured as an indication of NF-B activity.
- 206 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
Acetylcholine inhibit the production of IL-8 via mAchR activation
Since Ach and muscarine are able to inhibit the NF-B activity in Caco-2 cells, we
wanted to investigate whether cholinergic neurotransmitters inhibit NF-B-mediated
IL-8 production by Caco-2 cells. Therefore, we used Caco-2 cells that were
incubated for 20 minutes with increased concentrations of Ach, nicotine or
muscarine and after 3 hours we measured the IL-8 production in the supernatant.
Cells stimulated with Ach were also followed by incubation with 50nM neostigmine
for three hours. Neostigmine inhibits the degradation of acetylcholinesterase, so that
the Ach activity is prolonged. Besides neurones, also Caco-2 cells produce choline
acetyltransferases 37 and acetylcholinesterase 38, indicating that Caco-2 cells produce
and break-down Ach themselves.
Caco-2 cells that were incubated with Ach or muscarine significantly
produce less IL-8 in a concentration dependent manner compared to non-treated
cells, whereas increased concentrations of nicotine does not change the IL-8
secretion (see figure 2). Also 1000nM Ach treated Caco-2 cells followed by a 3-hour
incubation of 50nM neostigmine produce less IL-8 then non-treated cells (p<0.001).
- 207 -
Chapter 8
**
**
IL-8 production (%)
IL-8 production (%)
**
125
*
125
100
75
50
25
100
75
50
25
0
0
0
100
1000
a
0
100
10000
Acetylcholine (nM)
1000
10000
Acetylcholine (nM)
+ 50nM Neostigmine
b.
*
**
125
c.
IL-8 production (%)
IL-8 production (%)
125
100
75
50
25
0
**
0
100
1000
Nicotine (nM)
100
75
50
25
0
10000
0
d.
100
1000
10000
Muscarine (nM)
Figure 2. Caco-2 cells were incubated for 20 minutes with 0, 100, 1000 or 10.000 nM Ach (a. and
b.), nicotine (c.) or muscarine (d.). Three hours after this incubation, supernatant was collected and
measured for IL-8 production. Figure b. shows the effect of 20 minutes incubation of Ach followed
by three hours incubation of 50 nM neostigmine.
- 208 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
The production of IL-8 by Caco-2 cells is increased under influence of
several cytokines such as TNF- and IL-1. To test whether also Ach, nicotine and
muscarine inhibit cytokine-induced IL-8 production, Caco-2 cells were incubated
with increased concentrations of Ach, nicotine or muscarine for 20 minutes followed
by a 3 hour incubation of 1 or 100ng/ml TNF-or 10 or 25ng/ml IL-1. Thereafter
the IL-8 production was measured in the supernatant. Mainly Ach and muscarine
and not nicotine are able to inhibit significantly IL-8 production induced by low
doses of TNF- (1ng/ml), indicating that Ach acts via the mAchR on Caco-2 cells
(see figure 3). This effect is exacerbated when Caco-2 cells are also incubated with
neostigmine after Ach stimulation, probably because of the slowed break-down of
intrinsic produced Ach. In case of high doses of TNF-(100ng/ml), muscarine
cannot inhibit the IL-8 production.
Ach, nicotine and muscarine significantly inhibit IL-8 production induced
by IL-1, although it seems that Ach prefers to act via nAchR when IL-1
concentrations are low (10ng/ml), since also 100 and 1000nM nicotine significantly
decreases IL-8 production (p<0.01), whereas during high concentrations of IL-1
(25ng/ml) Ach acts via the mAchRs since then muscarine significantly decreases IL8 production in a concentration-dependent manner (see figure 4). Probably, this
inhibition of IL-1-induced IL-8 production is dependent on a short action of Ach,
because addition of neostigmine is less effective in inhibiting IL-8 production.
- 209 -
Chapter 8
1ng/ml TNF-
**
*
***
IL-8 production (%)
IL-8 production (%)
*
125
125
100
75
50
25
100
75
50
25
0
0
0
100
1000
Acetylcholine (nM)
a.
0
10000
100
1000
10000
Acetylcholine (nM)
+ 50nM Neostigmine
b.
***
125
IL-8 production (pg/ml)
IL-8 production (pg/ml)
***
*
100
75
50
25
0
0
100
1000
125
100
75
50
25
0
10000
Nicotine (nM)
c.
***
0
100
1000
10000
Muscarine (nM)
d.
100ng/ml TNF-
**
**
**
125
IL-8 production (%)
125
IL-8 production (%)
*
*
100
75
50
25
0
100
1000
50
25
e.
IL-8 production (pg/ml)
100
75
50
25
0
100
1000
10000
Nicotine (nM)
h.
100
1000
10000
Acetylcholine (nM)
+ 50nM Neostigmine
f.
125
0
0
10000
Acetylcholine (nM)
IL-8 production (pg/ml)
75
0
0
g.
100
125
100
75
50
25
0
0
100
1000
10000
Muscarine (nM)
Figure 3. Caco-2 cells were incubated for 20 minutes with 0, 100, 1000 or 10.000 nM Ach (a., b.,
e. and f.), nicotine (c. and g.) or muscarine (d. and h.) followed by 1ng/ml (a., b., c. and d.) or
100ng/ml TNF- incubation (e., f., g. and h.). Three hours after this incubation, supernatant was
collected and measured for IL-8 production. The figures b. and f. show the effect of 20 minutes
incubation of Ach followed by three hours incubation of 50 nM neostigmine together with 1ng/ml
or 100ng/ml TNF- respectively.
- 210 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
10ng/ml IL-1
*
**
**
IL-8 production (%)
IL-8 production (%)
100
75
50
25
100
75
50
25
0
0
0
100
1000
0
10000
Acetylcholine (nM)
a.
*
125
125
100
1000
10000
Acetylcholine (nM)
+ 50nM Neostigmine
b.
IL-8 production (pg/ml)
IL-8 production (pg/ml)
*
*
125
100
75
50
25
0
0
100
1000
10000
Nicotine (nM)
c.
125
100
75
50
25
0
0
100
1000
10000
Muscarine (nM)
d.
25ng/ml IL-1
**
125
IL-8 production (%)
IL-8 production (%)
125
100
75
50
25
100
75
50
25
0
0
0
100
1000
Acetylcholine (nM)
e.
0
100
10000
1000
10000
Acetylcholine (nM)
+ 50nM Neostigmine
f.
**
g.
*
125
IL-8 production (pg/ml)
IL-8 production (pg/ml)
**
100
75
50
25
0
0
100
1000
100
75
50
25
0
10000
Nicotine (nM)
125
h.
0
100
1000
10000
Muscarine (nM)
Figure 4. Caco-2 cells were incubated for 20 minutes with 0, 100, 1000 or 10.000 nM Ach (a., b.,
e. and f.), nicotine (c. and g.) or muscarine (d. and h.) followed by 10ng/ml (a., b., c. and d.) or
25ng/ml IL-1 incubation (e., f., g. and h.). Three hours after this incubation, supernatant was
collected and measured for IL-8 production. The figures b. and f. show the effect of 20 minutes
incubation of Ach followed by three hours incubation of 50 nM neostigmine together with 10ng/ml
or 25ng/ml IL-1 respectively.
- 211 -
Chapter 8
Acetylcholine reduces the permeability of Caco-2 monolayers
It has been described that several cytokines increase the intestinal epithelial
permeability via activation of the NF-B pathway
17-19
. To investigate whether Ach
influences the intestinal epithelial permeability, Caco-2 cells cultured on transwells
were incubated with different concentrations of Ach with or without 50nM
neostigmine followed by a cytokine mix which contains IL-1, TNF- and IFN-
for 72 hour. After 72 hours the TEER was measured. Moreover, 4kDa FITC dextran
particles were added at the mucosal site of the Caco-2 monolayer and the flux
though the monolayer was measured after 30 and 60 minutes.
The TEER is decreased under influence of the cytokine mix correlating
with an increase of 4kDa FITC dextran flux (see figure 5). Ach in the presence or
absence of neostigmine did not change the TEER. However, Ach increases the
dextran flux in a concentration-dependent manner, but in the presence of the
cytokine mix, Ach decreases the dextran flux decreased after 60 minutes.
Neostigmine has only an effect during the first 30 minutes of dextran flux.
- 212 -
140
130
120
110
100
90
80
70
60
50
40
30
20
10
0
0
10
10 0
00
0
10
10 0
00
0
10
10 0
00
Ach
Ach and neostigmine
Ach
Ach and neostigmine
0
10
10 0
00
%TEER
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
cytokine mix
Concentration Ach (nM)
a.
Flux (pmol/cm2/h)
200
Ach
Ach and neostigmine
Ach
Ach and neostigmine
150
100
50
0
10
10 0
00
0
10
10 0
00
0
10
10 0
00
0
10
10 0
00
0
cytokine mix
Concentration Ach (nM)
b.
Ach
Ach and neostigmine
Ach
Ach and neostigmine
150
100
50
0
10
10 0
00
0
10
10 0
00
0
10
10 0
00
0
-50
0
10
10 0
00
Flux (pmol/cm2/h)
200
cytokine mix
Concentration Ach (nM)
c.
Figure 5. TEER and 4kDa dextran flux measurements. After 72 hour of incubation with or without
cytokine mix and different concentrations of Ach in the presence or absence of 50nM neostigmine,
the TEER was measured (a.) and 4kDa FITC dextran particles were added at the mucosal site of
the Caco-2 monolayer and the flux through the monolayer was measured after 30 (b.) and 60
minutes (c.).
Finally, we investigated whether Ach influences the expression of the TJ
proteins occludin and ZO-1. After a 3 hour incubation of IL-1 and TNF-, there is
no translocation of occludin from the membrane to the cytosol under influence of
different concentrations of Ach or muscarine (see figure 6). After 72h, the cytokine
mix (IL-1, TNF- and IFN-) decreases the expression of ZO-1 in Caco-2 cells
compared to the untreated cells (see figure 7). The expression of ZO-1 is not
influenced by 100 or 1000nM Ach. However, when Caco-2 cells are treated with the
cytokine mix, 100 and 1000nM Ach restore the ZO-1 expression.
- 213 -
Chapter 8
Cytosol
Membrane
Ach
0
100
1000
Muscarine
10000
0
100
Ach
1000 10000
0
100
1000 10000
Muscarine
0
100
1000 10000
nM
Occludin
Actin
a.
Without cytokine mix
With cytokine mix
b.
c.
0nM Ach
d.
e.
100nM Ach
f.
g.
1000nM Ach
Figure 6. Expression of occludin and ZO-1. After 3 hours of incubation of IL-1 and TNF-, there
is no translocation of occludin from the membrane to the cytosol under influence of different
concentrations of Ach or muscarine (a.). After 72h, the cytokine mix (IL-1, TNF- and IFN-)
decreases the expression of ZO-1 in Caco-2 cells (c.) compared to the untreated cells (b.). The
expression of ZO-1 is not influenced by 100 or 1000nM Ach (d. and f.). However, when Caco-2
cells are treated with the cytokine mix, 100 and 1000nM Ach restore the ZO-1 expression (e. and
g.).
- 214 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
Electrical vagal nerve stimulation enhances transepithelial transport
under healthy conditions
Our in vitro data show that under inflammatory conditions Ach protects against
epithelial barrier dysfunction. To investigate whether Ach also changes epithelial
permeability under healthy conditions, the vagus nerve of mice was electrical
stimulated after ligation of the distal part of the ileum. FITC-labelled E. faecium
were injected in the ligated part of the ileum and after 30 minutes the mice were
sacrificed so that the ileum could be analysed for translocation of E. faecium to the
lamina propria and could be stained for lysomal LAMP-2 to see whether there is colocalisation.
It seems that under healthy conditions stimulation of the vagus nerve
increases paracellular transport of E. faecium compared to mice without VNS, since
more FITC-labelled E. faecium was present in the lamina propria of VNS-treated
mice (see figure 7). The uptake of E. faecium is not via lysosomes, because there is
no co-localisation of E. faecium with LAMP-2.
a.
b.
Figure 7. The vagus nerve of mice was electrical stimulated after ligation of the distal part of the
ileum where FITC-labelled E. faecium (green) was injected (b.). Moreover, ileum was stained for
lysosmal LAMP-2 (red). Mice without vagal nerve stimulation (VNS) were used as a control (a.).
- 215 -
Chapter 8
Discussion
TNF-
IL-1
The aim of this study was to investigate the
effect of Ach on the intestinal epithelial
Ach
barrier integrity. We demonstrated that Ach
Muscarine
Nicotine
NF-B
reduces TNF- or IL-1 induced IL-8
production of Caco-2 cells. Ach acts mainly
via the mAchR, since only muscarine inhibits
IL-8
the IL-8 production and not nicotine, although
nicotine also inhibits IL-8 production when it
Figure 8. Both Ach as muscarine
inhibit IL-8 production by Caco-2 cells,
whereas nicotine only inhibits IL-8
is induced by low concentrations of IL-1 (see
figure 8). Nevertheless, only Ach and
production when it is induced by low
muscarine and not nicotine reduce IL-1
concentrations of IL-1. Moreover,
induced NF-B activity of Caco-2 cells.
only Ach and muscarine reduce IL-1
Furthermore, we demonstrated that Ach
induced NF-B activity. Therefore, Ach
inhibits IL-8 production by Caco-2 cells
through mAchRs.
reduces 4kDa FITC Dextran flux though
Caco-2 monolayers and that Ach restores ZO1 expression of Caco-2 cells after cytokine
exposure. Our data implicate that Ach enforces the cellular processes leading to
increased permeability and dendritic cell-mediated antigen uptake and drainage to
the lymph nodes during homeostatic conditions, whereas under inflammatory
conditions, Ach protects against cytokine-induced enhanced permeability to prevent
excess exposure of the intestinal mucosa to luminal antigens and microbes.
It has been shown before that under inflammatory conditions activation of
the vagus nerve improves the intestinal barrier integrity. In a rat model of
haemorrhagic shock, a high fat diet which activates the vagus nerve via
cholecystekinin, significantly reduces bacterial translocation, HRP permeability and
rearrangement of ZO-1 39. Besides the improvement of the epithelial barrier integrity,
vagus nerve activation reduces local and systemic inflammation
39,40
. Moreover
intracerebroventricular injection of ghrelin in a rat sepsis and ischemia-reperfusion
- 216 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
model results in a reduction of intestinal epithelial permeability and bacterial
translocation
41,42
. Since vagotomy inhibits the effects of grhelin, this indicates that
ghrelin improves mucosal barrier integrity through the vagus nerve.
Cameron and Perdue showed that transcellular transport is increased via the
M3 muscarinic receptor under steady state conditions 27. Nevertheless, mice that are
deficient for the M3 muscarinic receptor are more susceptible for DSS-colitis
43
.
Although it has been described that the M3 muscarinic receptor is involved in
epithelial ion transport 44-46, these M3 muscarinic receptor-deficient mice developed
compensatory pathways. However, the colitis in these mice was characterised by the
presence of inflammation in the ileum, whereas colitis normally is restricted to the
colon and caecum. This indicates that the M3 muscarinic receptor has also antiinflammatory capacities. Our data also demonstrate that muscarine is able to
decrease NF-B activity and IL-8 production by Caco-2 cells.
McGilligan et al. demonstrated that nicotine decreases epithelial gut
permeability in Caco-2 cells 47. Moreover, not only M3 muscarinic-deficient mice,
but also 5 nAchR subunit-deficient mice are more susceptible for the development
of colitis
48
. We could only find decreased NF-B activity and IL-8 production of
Caco-2 cells by Ach and muscarine, indicating that this effect is mediated via
mAchRs. Probably, there is an effect of nicotine on the epithelial barrier integrity in
Caco-2 cells, but not on the NF-B activation and IL-8 production. That 5 nAchR
subunit-deficient mice are more susceptible for the development of colitis may also
be caused by an indirect effect on epithelial cells via interactions with enteric
neurons and glial cells, which also influences epithelial barrier integrity.
Most likely, the vagus nerve does not interact directly with intestinal
epithelial cells, but through enteric neurons and EGCs. EGCs produce Snitroglutathione, which plays an important role in the homeostasis of the epithelial
barrier. S-nitrosoglutathione is able to restore mucosal barrier function in colonic
biopsies from CD patients
49
. Ablation of jenunal and ileal glial cells results in
- 217 -
Chapter 8
inflammation of the intestine similar to IBD and reduces the intestinal barrier
integrity by altering expression of perijunctional F-actin and association of ZO-1
and occludin with the actin-cytoskeleton 49. Moreover, EGCs were able to strongly
inhibit intestinal epithelial cell proliferation in part by their release of transforming
growth factor-β1 (TGF-β1) 50. Interactions between the vagus nerve and EGCs seem
to be important to maintain the epithelial barrier integrity under healthy conditions.
However, also EGCs respond to inflammation by the production of IL-1 and TNF resulting in an increased epithelial permeability and activated immune cells
51
.
Probably, EGCs increase the epithelial permeability so that luminal antigens can
enter the lamina propria where immune cells will take up and process these antigens
to induce an inflammatory reaction. The release of Ach by enteric neurons or even
epithelial cells themselves may inhibit this inflammation.
Together, our data on the potential of vagal motor activity to affect barrier
function may be interpreted as a dual immune-supportive effect; enhancing immune
surveillance and supporting tolerance under healthy immune homeostasis, while
acting protective on inflammatory cytokine exposed epithelia. Firmly establish this
hypothesis more data are required that pin-point the in vivo role of vagal nerve
activity in support of gut immune homeostasis.
- 218 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
References
1 DeMeo,M.T. et al. (2002) Intestinal permeation and gastrointestinal disease. J. Clin. Gastroenterol. 34,
385-396
2 Benard,A. et al. (1996) Increased intestinal permeability in bronchial asthma. J. Allergy Clin. Immunol.
97, 1173-1178
3 Munkholm,P. et al. (1994) Intestinal permeability in patients with Crohn's disease and ulcerative colitis
and their first degree relatives. Gut 35, 68-72
4 Bjarnason,I. et al. (1995) Intestinal permeability: an overview. Gastroenterology 108, 1566-1581
5 Madara,J.L. (1987) Intestinal absorptive cell tight junctions are linked to cytoskeleton. Am. J. Physiol
253, C171-C175
6 Nusrat,A. et al. (2000) Molecular physiology and pathophysiology of tight junctions. IV. Regulation of
tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am. J. Physiol
Gastrointest. Liver Physiol 279, G851-G857
7 Tsukita,S. et al. (2001) Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2, 285-293
8 Furuse,M. et al. (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell
Biol. 123, 1777-1788
9 Furuse,M. et al. (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions
with no sequence similarity to occludin. J. Cell Biol. 141, 1539-1550
10 Martin-Padura,I. et al. (1998) Junctional adhesion molecule, a novel member of the immunoglobulin
superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol.
142, 117-127
11 Fanning,A.S. et al. (1998) The tight junction protein ZO-1 establishes a link between the
transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 273, 29745-29753
12 Wittchen,E.S. et al. (1999) Protein interactions at the tight junction. Actin has multiple binding
partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J. Biol. Chem. 274, 35179-35185
13 Bruewer,M. et al. (2003) Proinflammatory cytokines disrupt epithelial barrier function by apoptosisindependent mechanisms. J. Immunol. 171, 6164-6172
14 Madara,J.L. and Stafford,J. (1989) Interferon-gamma directly affects barrier function of cultured
intestinal epithelial monolayers. J. Clin. Invest 83, 724-727
15 Mullin,J.M. et al. (1992) Modulation of tumor necrosis factor-induced increase in renal (LLC-PK1)
transepithelial permeability. Am. J. Physiol 263, F915-F924
16 Zolotarevsky,Y. et al. (2002) A membrane-permeant peptide that inhibits MLC kinase restores barrier
function in in vitro models of intestinal disease. Gastroenterology 123, 163-172
- 219 -
Chapter 8
17 Ma,T.Y. et al. (2004) TNF-alpha-induced increase in intestinal epithelial tight junction permeability
requires NF-kappa B activation. Am. J. Physiol Gastrointest. Liver Physiol 286, G367-G376
18 Al-Sadi,R. et al. (2008) Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction
permeability. J. Immunol. 180, 5653-5661
19 Al-Sadi,R.M. and Ma,T.Y. (2007) IL-1beta causes an increase in intestinal epithelial tight junction
permeability. J. Immunol. 178, 4641-4649
20 Cario,E. (2008) Therapeutic impact of toll-like receptors on inflammatory bowel diseases: a multipleedged sword. Inflamm. Bowel. Dis. 14, 411-421
21 Cario,E. (2003) Toll-like receptors and intestinal defence: molecular basis and therapeutic implications.
Expert. Rev. Mol. Med. 5, 1-15
22 Ruhl,A. (2005) Glial cells in the gut. Neurogastroenterol. Motil. 17, 777-790
23 Lubbers,T. et al. (2009) Lipid-rich enteral nutrition reduces postoperative ileus in rats via activation of
cholecystokinin-receptors. Ann. Surg. 249, 481-487
24 Greenwood,B. and Mantle,M. (1992) Mucin and protein release in the rabbit jejunum: effects of
bethanechol and vagal nerve stimulation. Gastroenterology 103, 496-505
25 Neunlist,M. et al. (2003) Human ENS regulates the intestinal epithelial barrier permeability and a tight
junction-associated protein ZO-1 via VIPergic pathways. Am. J. Physiol Gastrointest. Liver Physiol 285,
G1028-G1036
26 van der Zanden,E.P. et al. (2009) Vagus nerve activity augments intestinal macrophage phagocytosis
via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology 137, 1029-39, 1039
27 Cameron,H.L. and Perdue,M.H. (2007) Muscarinic acetylcholine receptor activation increases
transcellular transport of macromolecules across mouse and human intestinal epithelium in vitro.
Neurogastroenterol. Motil. 19, 47-56
28 Ramakers,J.D. et al. (2007) Arachidonic acid but not eicosapentaenoic acid (EPA) and oleic acid
activates NF-kappaB and elevates ICAM-1 expression in Caco-2 cells. Lipids 42, 687-698
29 de Jonge,W.J. et al. (2005) Stimulation of the vagus nerve attenuates macrophage activation by
activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 6, 844-851
30 de Jonge,W.J. et al. (2005) Stimulation of the vagus nerve attenuates macrophage activation by
activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 6, 844-851
31 Ghia,J.E. et al. (2006) The vagus nerve: a tonic inhibitory influence associated with inflammatory
bowel disease in a murine model. Gastroenterology 131, 1122-1130
32 Luyer,M.D. et al. (2005) Nutritional stimulation of cholecystokinin receptors inhibits inflammation via
the vagus nerve. J. Exp. Med. 202, 1023-1029
33 Mioni,C. et al. (2005) Activation of an efferent cholinergic pathway produces strong protection against
myocardial ischemia/reperfusion injury in rats. Crit Care Med. 33, 2621-2628
- 220 -
Ach protects against inflammatory cytokine induced epithelial barrier dysfunction
34 The,F.O. et al. (2007) Activation of the cholinergic anti-inflammatory pathway ameliorates
postoperative ileus in mice. Gastroenterology 133, 1219-1228
35 van Westerloo,D.J. et al. (2006) The vagus nerve and nicotinic receptors modulate experimental
pancreatitis severity in mice. Gastroenterology 130, 1822-1830
36 van Westerloo,D.J. et al. (2005) The cholinergic anti-inflammatory pathway regulates the host
response during septic peritonitis. J. Infect. Dis. 191, 2138-2148
37 Cheng,K. et al. (2008) Acetylcholine release by human colon cancer cells mediates autocrine
stimulation of cell proliferation. Am. J. Physiol Gastrointest. Liver Physiol 295, G591-G597
38 Plageman,L.R. et al. (2002) Characterization of acetylcholinesterase in Caco-2 cells. Exp. Biol. Med.
(Maywood. ) 227, 480-486
39 de Haan,J.J. et al. (2008) Postshock intervention with high-lipid enteral nutrition reduces inflammation
and tissue damage. Ann. Surg. 248, 842-848
40 Luyer,M.D. et al. (2005) Nutritional stimulation of cholecystokinin receptors inhibits inflammation via
the vagus nerve. J. Exp. Med. 202, 1023-1029
41 Wu,R. et al. (2009) Orexigenic hormone ghrelin ameliorates gut barrier dysfunction in sepsis in rats.
Crit Care Med. 37, 2421-2426
42 Wu,R. et al. (2008) Orexigenic hormone ghrelin attenuates local and remote organ injury after
intestinal ischemia-reperfusion. PLoS. One. 3, e2026
43 Hirota,C.L. and McKay,D.M. (2006) M3 muscarinic receptor-deficient mice retain bethanecholmediated intestinal ion transport and are more sensitive to colitis. Can. J. Physiol Pharmacol. 84, 11531161
44 Chandan,R. et al. (1991) Cholinergic neurons and muscarinic receptors regulate anion secretion in pig
distal jejunum. Eur. J. Pharmacol. 193, 265-273
45 Dickinson,K.E. et al. (1992) Activation of T84 cell chloride channels by carbachol involves a
phosphoinositide-coupled muscarinic M3 receptor. Eur. J. Pharmacol. 225, 291-298
46 O'Malley,K.E. et al. (1995) Cholinergic activation of Cl- secretion in rat colonic epithelia. Eur. J.
Pharmacol. 275, 83-89
47 McGilligan,V.E. et al. (2007) The effect of nicotine in vitro on the integrity of tight junctions in Caco2 cell monolayers. Food Chem. Toxicol. 45, 1593-1598
48 Orr-Urtreger,A. et al. (2005) Increased severity of experimental colitis in alpha 5 nicotinic
acetylcholine receptor subunit-deficient mice. Neuroreport 16, 1123-1127
49 Savidge,T.C. et al. (2007) Enteric glia regulate intestinal barrier function and inflammation via release
of S-nitrosoglutathione. Gastroenterology 132, 1344-1358
50 Neunlist,M. et al. (2007) Enteric glia inhibit intestinal epithelial cell proliferation partly through a
TGF-beta1-dependent pathway. Am. J. Physiol Gastrointest. Liver Physiol 292, G231-G241
51 Ruhl,A. (2005) Glial cells in the gut. Neurogastroenterol. Motil. 17, 777-790
- 221 -
Chapter 8
- 222 -
Chapter
9.
Summary and discussion
Nederlandse samenvatting
Chapter 9
- 224 -
Summary and discussion/ Nederlandse samenvatting
Summary and discussion
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative
colitis (UC), are chronic inflammatory diseases of the intestine with an unknown
aetiology. Although the pathogenesis of these diseases is not well understood,
several components of the bacterial flora, the epithelial barrier, the immune system,
the nervous system and mutations in genes that are a part of these components have
been shown to play an essential role in the irregular nature of mucosal inflammation.
In this thesis we have investigated different parts of the pathogenesis of IBD.
The first part of this thesis describes the role of apoptosis in IBD (Chapter
2) and evaluates a new TNF- inhibitor in two different colitis models (Chapter 3).
Evidence is increasing that a defect in apoptosis is involved in the pathogenesis of
IBD. CD seems to be the cause of an intrinsic defect in the apoptotic pathway of
(autoreactive) T cells, resulting in excessive T cell responses. In UC, however, an
increased rate of apoptosis of epithelial cells is more likely involved in the
pathogenesis. Several therapeutic approaches in IBD induce apoptosis of
autoreactive T cells, such as sulphasalazine, azathioprine and infliximab. Infliximab
is a common used drug in CD, which induces apoptosis of T cells via the
transmembrane form of TNF-. Since infliximab has many side-effects, companies
are still developing new strategies to neutralise TNF-. In the first part of this thesis,
we describe the role of apoptosis in IBD (Chapter 2) and subsequently tested a new
inhibitor of soluble TNF-, based on the heavy chains of camel antibodies, the socalled nanobodies, in an acute and a chronic colitis mouse model (Chapter 3).
Unfortunately, these nanobodies did not ameliorate colitis, suggesting that the
transmembrane, and not the soluble form of TNF- has to be targeted
In the second part of this thesis we focused on the role of dendritic cells
(DC). DCs are key players in the innate and adaptive immune response and are
generally accepted to play a major role in the pathophysiology of CD. DCs located
in the lamina propria of the intestine migrate to the mesenteric lymph nodes (MLNs)
- 225 -
Chapter 9
where they present antigens to T cells. A number of different markers are described
to define DC populations. We have investigated which DC populations are present
in the colon and MLNs of CD patients by immunohistochemistry. It seems that there
are three different subpopulations of myeloid DCs present in colonic mucosa and
MLN of non-CD and CD patients (Chapter 4). These populations consist of the
following: (1) immature DCs or macrophages that express DC-SIGN, (2) mature
DCs that express S-100 or CD83, and (3) mature DCs that express BDCA3. BDCA1
and CD1a expressing DCs were virtually absent in the colon as well as in MLNs.
Immature DCs and macrophages are mainly localised at antigen-capturing sites such
as the mucosa and medullary cords, whereas mature DCs are present where antigen
is presented, including the T cell areas in colonic lymph follicles and MLNs. All
different DC markers give variable staining patterns so there is no marker for the
DC. Since DC-SIGN+ DCs were increased in the mucosa of CD patients, we wanted
to investigate whether polymorphisms in DC-SIGN are associated with IBD
(Chapter 5). Moreover, previous studies showed that mutations in NOD2, which is a
pattern recognition receptor similar to DC-SIGN, are associated with CD. Besides,
DC-SIGN is located in a susceptibility locus, like three other C-type lectins: MGL,
LLT1 and DCIR. Only polymorphisms in LLT1 seem to be associated with CD.
This is interesting since LLT1 is a ligand for CD161 and as a complex it inhibits NK
cell-mediated cytotoxicity and cytokine production. CD161 is a new surface marker
for human IL-17 producing Th17 cells. The Th17 phenotype has recently been
linked to CD by the fact that IL-22, IL-17 and IL-23 receptor levels are increased in
CD.
The last section of this thesis describes how the enteric nervous system
influences barrier function of the intestine (Chapter 6 and 8) and we investigated
whether two new 7 nAchR agonists can ameliorate experimental colitis (Chapter
7). Besides the anti-inflammatory capacities of acetylcholine (Ach), evidence is
accumulating that Ach also plays an important role in the homeostasis of the
epithelial barrier integrity. Ach can signal via nicotinic or muscarinic acetylcholine
receptors (nAchRs or mAchRs). We tested two selective 7 nAchR agonists
- 226 -
Summary and discussion/ Nederlandse samenvatting
(ARR17779,
(-)-spiro[1-azabicyclo[2.2.2]
octane-3,5′-oxazolidin-2′-one
and
GSK1345038A) on disease severity in two mouse models of experimental colitis.
Both 7 nAchR agonists worsened colitis or were ineffective in these colitis models,
questioning the further development of 7 nAchR agonists as treatment for colitis.
In the last study we investigated the effect of Ach, nicotine and muscarine on the
epithelial barrier function (Chapter 8). We showed that Ach and muscarine, but not
nicotine, inhibit IL-8 production of Caco-2 cells induced by TNF- and IL-1,
indicating that Ach mainly acts via the mAchRs. Interestingly, low concentrations of
nicotine also inhibited IL-8 production induced by low concentrations of IL-1.
Nevertheless, only Ach and muscarine and not nicotine reduce IL-1 induced NFB activity of Caco-2 cells. Furthermore, we demonstrated that Ach reduces 4kDa
dextran flux though Caco-2 monolayers and restores ZO-1 expression of Caco-2
cells after cytokine exposure. However, in healthy mice, Ach increases bacterial
translocation. These data indicate that Ach protects epithelial cells from the
detrimental effects of IL-1 and TNF- on the integrity of the intestinal epithelial
barrier via activation of mAchRs, but that under healthy conditions Ach increases
epithelial permeability.
Since we showed in two studies that promising new therapies, a TNF-
inhibitor and specific 7 nAchR agonists, did not work in different mice models, it
is important that future research will focus more at IBD pathology at molecular
level. This means that we have to elucidate more about the role of soluble and
transmembrane TNF- in inflammation, because the different forms of TNF- seem
to have another function. In addition, it seems crucial to unravel the mechanisms
controlling the production of TNF-, in other words, what are the triggers for a cell
to produce transmembrane TNF- and which signals are responsible for the
cleavage of transmembrane TNF- into its soluble form, and perhaps most
importantly, do these signals differ between IBD patients and healthy subjects. This
knowledge will be of crucial importance to develop strategies to interfere with these
- 227 -
Chapter 9
mechanisms and/or to develop new anti-TNF therapies that are more efficient and
devoid side-effects.
Although our 7 nAchR agonists did not ameliorate colitis in mice, these
findings do not exclude that activation of other nAchRs may have therapeutic effects
as the anti-inflammatory effect of vagal nerve stimulation may result from
interaction with other nAchRs. Besides, not only nAchRs could be a potential target
in the treatment of IBD and other intestinal diseases, but also mAchRs since
activation of mAchRs decreases intestinal permeability and inhibits the NF-B
pathway under inflammatory conditions. We do not know, however, which mAchR
is involved in this processes, although the M3 muscarinic receptor may be a good
candidate. Therefore, future research should focus on the type of mAchRs expressed
by intestinal epithelial cells in situ and in vitro and whether activation of this
receptor can ameliorate colitis in mice models.
Besides more research at molecular level, it is also necessary to do more
research at cellular level. We demonstrated that there are several DC populations
present in the mucosa of the colon and MLNs, but the exact role of these DC
populations in the pathogenesis of IBD is not known yet. Especially, DC-SIGN+ and
s-100+ DCs are interesting as they are increased in IBD patients. The challenge thus
is to isolate these DC populations from the intestine and MLN for further in detail
study. Probably, it is not possible to culture those cell populations, but RNA and
protein isolation may give us some more information about the pathways, i.e. NOD2
and NALP3 signalling, that could be involved in the pathogenesis of IBD. Finally,
more genetic research is necessary to make a better patient profile. Not all IBD
patients have the same polymorphisms. Although the association of polymorphisms
in LTT1 with CD is not very strong, it is important to investigate whether the
combination of polymorphisms in LTT1 and other genes increases the risk to
develop CD. Future studies will have to evaluate which combination indeed leads to
an increased disease risk and should try to identify clusters of patients with different
combinations of polymorphisms in for example NOD2, NALP3 and TLR4 or in
- 228 -
Summary and discussion/ Nederlandse samenvatting
LLT1 and IL-23R. The final goal is to predict which treatment will be most effective
in these patients and whether family members have an increased risk to develop IBD
so that the disease could be prevented or the onset of the disease could be delayed.
In conclusion, our data demonstrate that many factors are involved in the
pathogenesis of IBD and that all these components could be targets in the treatment
of these diseases, although much more research is required to understand how all
these processes interact.
- 229 -
Chapter 9
- 230 -
Summary and discussion/ Nederlandse samenvatting
Nederlandse samenvatting
Inflammatoire darmziekten (IBD), waaronder de ziekte van Crohn (CD) en colitis
ulcerosa (UC), zijn chronische ziekten van de darm waarvan de oorzaak onbekend
is. Ondanks dat de pathogenese van deze ziekten niet goed wordt begrepen, zijn er
wel aanwijzingen dat de bacteriële flora, de epiheliale barrière, het immuunsysteem,
het zenuwstelsel en genetische aanleg een belangrijke rol spelen in het ontstaan
mucosale ontsteking. In dit proefschrift hebben wij verschillende aspecten van de
pathogenese van IBD onder de loep genomen.
Het eerste deel van dit proefschrift beschrijft de rol van celdood, ook wel
apoptose genoemd, in IBD (hoofdstuk 2) en evalueren we een nieuwe TNF-
remmer in twee verschillende colitismodellen in de muis (hoofdstuk 3). Er is steeds
meer bewijs dat een defect in apoptose betrokken is bij de pathogenese van IBD.
Een intrinsiek defect in de signaal transductie van (autoreactieve) T cellen wat leidt
tot verminderde apoptose van deze cellen, lijkt een oorzaak te zijn van CD. Echter,
in UC lijkt juist excessieve apoptose van epitheelcellen een belangrijke rol te spelen
in de pathogenese. Verschillende therapieën zijn erop gericht om apoptose van
autoreactieve T cellen te induceren, zoals sulphasalazine, azathioprine en infliximab.
Infliximab wordt algemeen gebruikt in de behandeling van CD en het induceert
apoptose van T cellen via transmembraan TNF-. Omdat infliximab veel
bijwerkingen heeft, ontwikkelen farmaceutische bedrijven tot op de dag van
vandaag nieuwe medicijnen om TNF- te neutraliseren. In het eerste deel van dit
proefschrift beschrijven we de rol van apoptose in IBD (hoofdstuk 2) en testen we
vervolgens een nieuwe remmer van de vrije vorm van TNF-. Deze TNF- remmer
is gebaseerd op de zware ketens van kameelantistoffen, de zogenaamde nanobodies,
en is getest in een acuut en een chronisch colitis muismodel (hoofdstuk 3). Helaas
hadden deze nanobodies geen effect op het ziekteverloop van colitis in deze
modellen, waarschijnlijk omdat niet de vrije vorm van TNF-, maar de membraangebonden vorm van TNF- moet worden geremd.
- 231 -
Chapter 9
In het tweede deel van dit proefschrift zoomen we in op de rol van
dendritische cellen (DCs). DCs spelen een belangrijke rol in aangeboren en
verworven immuunresponsen en het is algemeen aangenomen dat voor deze cellen
een belangrijke rol is weggelegd in de pathogenese van CD. DCs bevinden zich in
de lamina propria van de darm waar zij antigenen oppikken om die vervolgens te
presenteren
aan
lymfocyten
in
de
mesenterische
lymfeknopen
(MLNs).
Verschillende markers zijn beschreven om DCs te typeren. Wij hebben onderzocht
welke DC populaties er aanwezig zijn in de dikke darm en in de MLNs door middel
van immunohistochemie. Het lijkt erop dat er drie verschillende subpopulaties zijn
van myeloïde DCs in de mucosa van de dikke darm en MLNs van patiënten met en
zonder CD (hoofdstuk 4), namelijk (1) immature DCs of macrofagen die DC-SIGN
tot expressie brengen, (2) mature DCs die s-100 en/of CD83 tot expressie brengen
en (3) DCs die BDCA3 tot expressie brengen. BDCA1 en CD1a positieve DCs zijn
afwezig in de dikke darm en MLN. Immature DCs zijn voornamelijk gelokaliseerd
op plaatsen waar antigeen wordt opgenomen, zoals de lamina propria en de
medullaire strengen, terwijl mature DCs vooral aanwezig zijn op plaatsen waar
antigeen wordt gepresenteerd, inclusief de T cel gebieden in follikels in de dikke
darm en MLNs. Al deze verschillende markers zijn gelokaliseerd op verschillende
plaatsen in de dikke darm en MLNs wat aangeeft dat er geen specifieke marker is
voor dé DC. Omdat DC-SIGN+ DCs verhoogd aanwezig zijn in de mucosa van CD
patiënten wilden we onderzoeken of polymorfismen in DC-SIGN geassocieerd zijn
met IBD (hoofdstuk 5). Bovendien lieten eerdere studies al zien dat mutaties in
NOD2, dat evenals DC-SIGN een pattern recognition receptor is, zijn geassocieerd
met CD. Daarnaast is DC-SIGN samen met vele andere C-type lectins waartoe DCSIGN behoort, gelegen in zogenaamde susceptibility loci. Naast DC-SIGN
onderzochten we eveneens of polymorfismen in drie andere C-type lectins, namelijk
MGL, LLT1 en DCIR geassocieerd zijn met IBD. Alleen polymorfismen in LLT1
blijken geassocieerd te zijn met CD. Dit is interessant, omdat LLT1 een ligand is
voor CD161 en als een complex remt het de cytotoxiciteit van natural killer cellen
en cytokine productie. CD161 is een nieuwe oppervlakte marker voor humaan IL-17
- 232 -
Summary and discussion/ Nederlandse samenvatting
producerende Th17 cellen. Deze Th17 cellen zijn recent gelinked aan CD door het
feit dat IL-22, IL-17 en IL-23 levels in CD zijn verhoogd.
Het laatste deel van dit proefschrift beschrijft hoe het enterische
zenuwstelsel de barrière functie van de darm beïnvloedt (hoofdstuk 6 en 8) en testen
we twee nieuwe specifieke 7 nAchR agonisten in twee verschillende
colitismodellen (hoofdstuk 7). Naast de anti-inflammatoire werking van
acetylcholine (Ach), komt er steeds meer bewijs dat Ach ook een rol speelt in de
homeostase van de epitheliale barrière. Ach kan via nicotinerge of muscarinerge
receptoren (nAchRs en mAchRs) signaleren. We hebben twee selectieve 7 nAchR
agonisten, (ARR17779, (-)-spiro[1-azabicyclo[2.2.2] octane-3,5′-oxazolidin-2′-one
and GSK1345038A) getest in twee colitis muismodellen. Beide agonisten
verslechteren colitis of zijn ineffectief. Dat zet vraagtekens bij het verder
ontwikkelen van 7 nAchR agonisten als behandeling voor colitis. In de laatste
studie hebben we het effect van Ach, nicotine en muscarine op de epitheliale
barrière functie onderzocht (hoofdstuk 8). We laten zien dat Ach en muscarine, maar
niet nicotine, de IL-8 productie van Caco-2 cellen remt welke is geïnduceerd door
TNF- en IL-1. Dit geeft aan dat Ach voornamelijk via mAchRs dit effect
induceert, hoewel bij lage concentraties van IL-1, nicotine wel de IL-8 productie
remt. Niettemin kunnen alleen Ach en muscarine de NF-B pathway in Caco-2
cellen remmen. Verder laten we zien dat Ach de 4kDa FITC-Dextran flux door
Caco-2 monolagen reduceert onder inflammatoire condities en dat het ZO-1
expressie van Caco-2 cellen verbetert na expositie van een cytokine cocktail. Echter,
in gezonde muizen verhoogt Ach de bacteriële translocatie. Deze data laten zien dat
Ach de epitheliale barrière beschermt tegen de verhogende permeabiliteit van TNF en IL-1 via activatie van mAchRs, maar dat onder normale omstandigheden Ach
de epitheliale permeabiliteit verhoogt.
Omdat we in twee studies hebben laten zien dat een nieuwe TNF- remmer
en twee nieuwe specifieke 7 nAchR agonisten geen effect hebben op het
ziekteproces in verschillende colitismodellen in de muis is het belangrijk dat het
- 233 -
Chapter 9
onderzoek betreffende de IBD pathologie zich meer zal richten op moleculair
niveau. Dat betekent onder andere dat we meer zullen moeten kijken naar de rol van
het vrije en membraangebonden TNF- in ontsteking, omdat de verschillende
vormen van TNF- blijkbaar verschillende functies hebben. Bovendien is het
belangrijk om te onderzoeken welke mechanismen de productie van TNF-
beïnvloeden, met andere woorden: wat zijn de stimuli die maken dat een cel
membraangebonden TNF- gaat maken en welke signalen zijn verantwoordelijk
voor het afsplitsen van het vrije TNF-. Misschien is wel de meest belangrijke vraag
of er verschillen zijn in deze mechanismen tussen gezonde personen en IBD
patiënten. Deze kennis is onontbeerlijk om nieuwe therapieën te ontwikkelen die
efficiënter werken en minder bijwerkingen hebben.
Hoewel onze 7 nAchR agonisten niet werkzaam bleken in colitis
muismodellen kunnen we niet uitsluiten dat andere nAchRs verantwoordelijk
kunnen zijn voor het ontstekingsremmende effect van nervus vagus activatie.
Daarnaast kan ook de activatie van mAchRs als potentiële kandidaat in de
behandeling van IBD worden gezien, omdat activatie van mAchR resulteerde in een
daling van de intestinale permeabiliteit en een remming van de NF-B pathway. We
weten echter niet via welke mAchR deze effecten worden gemedieerd, hoewel de
M3 muscarinerge receptor een goede kandidaat lijkt te zijn. Daarom zal toekomstig
onderzoek zich moeten richten op welke typen van mAchRs tot expressie worden
gebracht door intestinale epitheelcellen in situ als wel in in vitro en of activatie van
deze receptoren daadwerkelijk resulteert in een verminderde ziekteactiviteit in colitis
muismodellen.
Naast meer onderzoek op moleculair niveau is het ook belangrijk om meer
onderzoek te doen op cellulair niveau. We hebben laten zien dat er verschillende DC
populaties aanwezig zijn in de mucosa van de dikke darm en de MLNs, maar de
exacte rol van deze DC populaties in IBD is niet duidelijk. Vooral de DC-SIGN+ en
s-100+ DCs zijn interessant, omdat deze meer aanwezig zijn bij CD patiënten. De
uitdaging is om deze DC populaties te isoleren uit de darm en MLN om ze meer in
- 234 -
Summary and discussion/ Nederlandse samenvatting
detail te kunnen bestuderen. Waarschijnlijk zal het niet mogelijk zijn om deze
celpopulaties in kweek te brengen, maar RNA en eiwit isolatie zal ons ongetwijfeld
de nodige informatie verschaffen over welke pathways betrokken kunnen zijn bij het
ziekteproces; te denken valt aan de NOD2 en NALP3 signalering. Tot slot is het
belangrijk om meer genetisch onderzoek te doen om een beter patiëntenprofiel te
krijgen; niet alle IBD patiënten hebben immers dezelfde polymorfismen. Ondanks
dat de associatie tussen LTT1 en CD niet erg sterk is, is het toch belangrijk om te
onderzoeken of de combinatie van polymorfismen in LTT1 en andere genen een
sterkere associatie zal opleveren. Toekomstige studies zullen moeten uitwijzen of
combinaties van polymorfismen in verschillende genen inderdaad leiden tot een
verhoogde kans om IBD te ontwikkelen. Zo zouden we bijvoorbeeld clusters van
patiënten kunnen identificeren die polymorfismen in NOD2, NALP3 en TLR4
hebben of juist in LTT1 en IL-23R. Het uiteindelijke doel is om te voorspellen
welke behandeling het meest effectief zal zijn en of familieleden een verhoogd risico
hebben om de ziekte te krijgen zodat we de ziekte kunnen voorkomen of kunnen
uitstellen. Kortom, de behandeling van de patiënt zal op maat gemaakt zijn.
In conclusie, deze data laten zien dat vele factoren zijn betrokken bij de
pathogenese van IBD en dat al deze componenten een target kunnen zijn in de
behandeling van deze ziekten, hoewel meer onderzoek nodig is om te begrijpen hoe
al deze processen met elkaar interacteren.
- 235 -
Chapter 9
- 236 -
Chapter
10.
Appendices:
Dankwoord
Curriculum Vitae
List of publications
Chapter 10
- 238 -
Appendices
Dankwoord
Natuurlijk maak je een proefschrift niet alleen, daar zijn altijd meerdere mensen bij
betrokken. Daarom schrijven de meeste promovendi een dankwoord achterin hun
proefschrift. Ik heb echter gekozen om aan een ieder die een bijdrage heeft geleverd
aan mijn proefschrift in welke vorm dan ook een persoonlijke boodschap te brengen.
Bovendien draag ik dit proefschrift op aan diezelfde mensen die mij geholpen
hebben bij het bedenken, uitvoeren en opschrijven van de experimenten, en aan de
mensen die mij de afgelopen jaren tot grote steun zijn geweest.
Marleen
- 239 -
Chapter 10
- 240 -
Appendices
Curriculum Vitae
Marleen Ingeborg Verstege is geboren op 18 september 1980 te Maarssen. Na het
behalen van haar Atheneum diploma aan de Rijksscholengemeenschap Broklede te
Breukelen in 1999, begint zij in hetzelfde jaar aan de studie medische biologie aan
de Universiteit van Amsterdam. Gedurende haar studietijd loopt Marleen stage bij
Vincent Christoffels en Antoon Moorman op de afdeling anatomie en embryologie
aan het Academisch Medisch Centrum (AMC) te Amsterdam, waar zij onderzoek
doet aan de embryonale ontwikkeling van het hart. Tijdens deze stage ontmoet zij
haar huidige vriend Peter Boorsma. In 2003 haalt Marleen eveneens een propedeuse
geneeskunde, maar zij besluit na drie jaar geneeskunde-onderwijs dat haar hart ligt
bij het laboratoriumwerk. Marleen vervolgt haar studie medische biologie met een
stage bij Anje te Velde en Daan Hommes op de afdeling experimentele inwendige
geneeskunde aan het AMC, waar zij onderzoek doet aan de pathogenese van
inflammatoire darmziekten. Tijdens deze stage zet Marleen bij NovImmune te
Genève in Zwitserland het TNBS-geïnduceerde colitis muizenmodel op. Haar laatste
stage loopt Marleen bij Elisabeth Hultgren-Hörnquist en Paul Bland op afdeling
klinische immunologie aan de Universitet Göteborg te Gothenburg in Zweden, waar
zij onderzoek doet naar de rol van tight junctions en probiotica bij inflammatoire
darmziekten. Marleen schrijft haar scriptie over immuuntherapie bij multiple
sclerose onder begeleiding van Eddy Wierenga en Martien Kapsenberg, verbonden
aan de afdeling celbiologie en histologie aan het AMC. In 2005 studeert Marleen
cum laude af en begint zij haar promotie-onderzoek bij Anje te Velde op de afdeling
experimentele inwendige geneeskunde, later omgedoopt tot het centrum voor
experimentele en moleculaire geneeskunde. Maria Rescigno, verbonden aan de
afdeling experimentele oncologie aan het European Institute of Oncology te Milaan
in Italië, nodigt Marleen uit om in haar laboratorium te leren hoe dendritische cellen
uit darmweefsel geïsoleerd kunnen worden. In haar tweede jaar zijn Wouter de
Jonge en Guy Boeckxstaens Marleen mede gaan begeleiden. Het promotieonderzoek betreft uiteenlopende onderwerpen waaronder de invloed van apoptose,
genen, immuuncellen en neurotransmitters op inflammatoire darmziekten waar dit
- 241 -
Chapter 10
boekje uit voort is gekomen. Op 22 december 2007 krijgen Marleen en Peter een
dochter, genaamd Lusanne Sofie. Gedurende de laatste maanden van haar promotieonderzoek is de vakgroep gastroenterologie ingetrokken bij het Levercentrum welke
is omgedoopt tot het Tytgat Instituut. Sinds september 2009 is Marleen werkzaam
als research analist bij Yvette van Kooyk en Wendy Unger, verbonden aan de
afdeling moleculaire celbiologie en immunologie aan het VU Medisch Centrum te
Amsterdam. Medio 2010 is het proefschrift gecomplementeerd en zal Marleen deze
gaan verdedigen op 29 juni. Zij is dan inmiddels zeven maanden zwanger van haar
tweede kind welke eind augustus wordt verwacht.
- 242 -
Appendices
List of publications
Wolfkamp SC, Verstege MI, Meisner S, Stokkers PC, te Velde AA. Single
nucleotide polymorphisms in C-type lectin genes, clustered in the IBD2 and IBD6
susceptibility loci, may play a role in the pathogenesis of inflammatory bowel
diseases. Manuscript in preparation.
Snoek SA, Verstege MI, van den Wijngaard RM, de Jonge WJ. The enteric nervous
system as regulator of intestinal epithelial barrier function in health and disease. Exp
Rev Gastroenterol Hepatol. 2010. Submitted.
Snoek SA, Verstege, MI, van der Zanden EP, Deeks N, Bulmer DC, Skynner M,
Lee K, te Velde AA, Boeckxstaens GE, de Jonge WJ. Selective 7 nicotinic
acetylcholine receptor agonists worsen disease in experimental colitis. Br J
Pharmacol. 2010 May;160(2):322-33.
van Zoelen MAD, Verstege MI, Draing C, de Beer R, van ’t Veer C, Florquin S,
Bresser P, van der Zee JS, te Velde AA, von Aulock S, van der Poll T. Endogenous
MCP-1 promotes lung inflammation induced by LPS and LTA. Submitted.
van Zoelen MAD, Florquin S, de Beer R, Pater JM, Verstege MI, Meijers JC, van
der Poll T. Urokinase plasminogen activator receptor-deficient mice demonstrate
reduced hyperoxia-induced lung injury. Am J Pathol. 2009 Jun;174(6):2182-9.
Ramakers JD, Mensink RP, Verstege MI, te Velde AA, Plat J. An arachidonic acidenriched diet does not result in more colonic inflammation as compared with fish
oil- or oleic acid-enriched diets in mice with experimental colitis. Br J Nutr. 2008
Aug;100(2):347-54.
Verstege MI, ten Kate FJ, Reinartz SM, van Drunen CM, Slors FJ, Bemelman WA,
Vyth-Dreese FA, te Velde AA. Dendritic cell populations in colon and mesenteric
lymph nodes of patients with Crohn's disease. J Histochem Cytochem. 2008
Mar;56(3):233-41.
Ramakers JD, Verstege MI, Thuijls G, Te Velde AA, Mensink RP, Plat J. The
PPARgamma agonist rosiglitazone impairs colonic inflammation in mice with
experimental colitis. J Clin Immunol. 2007 May;27(3):275-83.
Verstege MI, te Velde AA, Hommes DW. Apoptosis as a therapeutic paradigm in
inflammatory bowel diseases. Acta Gastroenterol Belg. 2006 Oct-Dec;69(4):406-12.
te Velde AA, Verstege MI, Hommes DW. Critical appraisal of the current practice
in murine TNBS-induced colitis. Inflamm Bowel Dis. 2006 Oct;12(10):995-9.
- 243 -
Chapter 10
- 244 -