Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Fischer–Tropsch process wikipedia , lookup
Oxidation state wikipedia , lookup
Cluster chemistry wikipedia , lookup
Metal carbonyl wikipedia , lookup
Spin crossover wikipedia , lookup
Metalloprotein wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
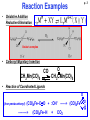
Organometallic Chemistry JHU Course 030.442 Prof. Kenneth D. Karlin Spring, 2009 Kenneth D. Karlin Department of Chemistry, Johns Hopkins University [email protected] http://www.jhu.edu/~chem/karlin/ Organometallic Chemistry p. 1 030.442 Prof. Kenneth D. Karlin Spring, 2009 Class Meetings: TTh, 12:00 – 1:15 pm Textbook – The Organometallic Chemistry of the Transition Metals” 4th Ed., R. H. Crabtree Course Construction: Homeworks, Midterm Exams (1 or 2), Oral Presentations Rough Syllabus Most or all of these topics • Introduction, History of the field • Reaction Types Oxidative Addition • Transition Metals, d-electrons Reductive elimination – • Bonding, 18 e Rule (EAN Rule) Insertion – Elimination Nucleophilic/electrophilic Rxs. • Ligand Types / Complexes • Types of Compounds • Catalysis – Processes Wacker oxidation M-carbonyls, M-alkyls/hydrides Monsanto acetic acid synthesis M-olefins/arenes Hydroformylation M-carbenes (alkylidenes alkylidynes) Polymerization- Olefin metathesis Water gas-shift reaction Other Fischer-Tropsch reaction p. 2 p. 3 Reaction Examples • Oxidative Addition Reductive Elimination Vaska’s complex • Carbonyl Migratory Insertion CH3Mn(CO)5 CO O CH3CMn(CO)5 • Reaction of Coordinated Ligands O (Iron pentacarbonyl) (CO)4Fe–C O + :OH– ––––> (CO)4Fe ––––––> (CO)4Fe–H + CO2 O H Reaction Examples - continued p. 4 • Wacker Oxidation C2H4 (ethylene) + ½ O2 –––> CH3CH(O) (acetaldehyde) Pd catalyst, Cu (co-catalyst) • Monsanto Acetic Acid Synthesis CH3OH (methanol) + CO –––> CH3C(O)OH (acetic acid) (Rh catalyst) • Ziegler-Natta catalysts – Stereoregular polymerization of 1-alkenes (a-olefins) 1963 Nobel Prize n CH2=CHR –––> –[CH2-CHR]n– Catalyst: Ti compounds and organometalllic Al compound (e.g., (C2H5)3Al ) • Olefin metathesis – variety of metal complexes 2005 Nobel Prize – Yves Chauvin, Robert H. Grubbs, Richard R. Schrock p. 5 Organo-transition Metal Chemistry History-Timeline • Main-group Organometallics 1760 - Cacodyl – tetramethyldiarsine, from Co-mineral with arsenic 1899 –> 1912 Nobel Prize: Grignard reagents (RMgX) n-Butyl-lithium • 1827 – “Zeise’s salt” - K+ [(C2H4)PtCl3]– Synthesis: PtCl4 + PtCl2 in EtOH, reflux, add KCl Bonding- Dewar-Chatt-Duncanson model p. 6 Organo-transition Metal Chemistry History-Timeline (cont.) 1863 - 1st metal-carbonyl, [PtCl2(CO)2] 1890 – L. Mond, (impure) Ni + xs CO –––> Ni(CO)4 (highly toxic) 1900 – M catalysts; organic hydrogenation (---> food industry, margerine) 1930 – Lithium cuprates, Gilman regent, formally R2Cu–Li+ 1951 – Ferrocene discovered. 1952 -- Sandwich structure proposed (Cp)2Fe Cp = cyclopentadienyl anion) (h5-C5H5)2Fe (pentahapto) Solid-state structure Ferrocene was first prepared unintentionally. Pauson and Kealy, cyclopentadieny-MgBr and FeCl3 (goal was to prepare fulvalene) But, they obtained a light orange powder of "remarkable stability.”, later accorded to the aromatic character of Cp– groups. The sandwich compound structure was described later; this led to new metallocenes chemistry (1973 Nobel prize, Wilkinson & Fischer). The Fe atom is assigned to the +2 oxidation state (Mössbauer spectroscopy). The bonding nature in (Cp)2Fe allows the Cp rings to freely rotate, as observed by NMR spectroscopy and Scanning Tunneling Microscopy. ----> Fluxional behavior. (Note: Fe-C bond distances are 2.04 Å). p. 7 Organo-transition Metal Chemistry History-Timeline (cont.) 1955 - Cotton and Wilkinson (of the Text) discover organometallic-complex fluxional behavior (stereochemical non-rigidity) The capability of a molecule to undergo fast and reversible intramolecular isomerization, the energy barrier to which is lower than that allowing for the preparative isolation of the individual isomers at room temperature. It is conventional to assign to the stereochemically non-rigid systems those compounds whose molecules rearrange rapidly enough to influence NMR line shapes at temperatures within the practical range (from –100 °C to +200 °C ) of experimentation. The energy barriers to thus defined rearrangements fall into the range of 5-20 kcal/mol (21-85 kJ/mol). Aside: Oxidation State 18-electron Rule p. 8 Fluxional behavior; stereochemical non-rigidity (cont.) Butadiene iron-tricarbonyl Xray- 2 CO’s equiv, one diff., If retained in solution, expect, 2:1 for 13-C NMR. But, see only 1 peak at RT. Cooling causes a change to the 2:1 ratio expected. Two possible explanations: (1)Dissociation and re-association or (2) rotation of the Fe(CO)3 moiety so that CO’s become equiv. Former seems not right, because for example addition of PPh3 does NOT result in substitution to give (diene)M(CO)2PPh3. Note: You can substitute PPh3 for CO, but that requires either high T or hv. So, the equivalency of the CO groups is due to rotation without bond rupture, pseudorotation. 13C-NMR spectra CO region, only p. 9 Berry Pseudorotation Pseudorotation: Ligands 2 and 3 move from axial to equatorial positions in the trigonal bipyramid whilst ligands 4 and 5 move from equatorial to axial positions. Ligand 1 does not move and acts as a pivot. At the midway point (transition state) ligands 2,3,4,5 are equivalent, forming the base of a square pyramid. The motion is equivalent to a 90° rotation about the M-L1 axis. Molecular examples could be PF5 or Fe(CO)5. p. 10 The Berry mechanism, or Berry pseudorotation mechanism, is a type of vibration causing molecules of certain geometries to isomerize by exchanging the two axial ligands for two of the equatorial ones. It is the most widely accepted mechanism for pseudorotation. It most commonly occurs in trigonal bipyramidal molecules, such as PF5, though it can also occur in molecules with a square pyramidal geometry. The process of pseudorotation occurs when the two axial ligands close like a pair of scissors pushing their way in between two of the equatorial groups which scissor out to accommodate them. This forms a square based pyramid where the base is the four interchanging ligands and the tip is the pivot ligand, which has not moved. The two originally equatorial ligands then open out until they are 180 degrees apart, becoming axial groups perpendicular to where the axial groups were before the pseudorotation.