Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Implicit solvation wikipedia , lookup
Rosetta@home wikipedia , lookup
Protein design wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Structural alignment wikipedia , lookup
List of types of proteins wikipedia , lookup
Protein moonlighting wikipedia , lookup
Circular dichroism wikipedia , lookup
Homology modeling wikipedia , lookup
Protein purification wikipedia , lookup
Trimeric autotransporter adhesin wikipedia , lookup
Protein mass spectrometry wikipedia , lookup
Protein folding wikipedia , lookup
Western blot wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Protein domain wikipedia , lookup
Protein–protein interaction wikipedia , lookup
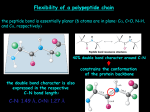
Announcement I am Hyun-Soo Cho, in Biology Department. This course is Biophysics, (1) How to get lecture slides structure.yonsei.ac.kr/ File name: psf_Ch1.ppt (2) Exam : 2 times (Mid, Final Exam) Problem types: Short or long answer 100% Place: Lecture Room 과S118A Posting of score in Exam: on the board at room SB134, 신과학원, Announcement (continued) (3) Assignment (homework): Please read your textbook before or/and after each class (4) Grading: Mid exam (35%) + Final exam (35%) + Presentation (15%) + attendance (5%) + Quiz (10%) (5) Participating in this Biophysics Course Thanks everyone for your interest on this class Have good manners : No cell phone (no message), No chatting I hope you would keep your honor during this course Announcement (continued) (6) Interviewing with me You must see me during this course at least one time My office hours: PM 5:00-5:30 on Wed How: First, Contact me by E-mail or telephone E-mail address: [email protected], 2123-5651 Protein Structure and Function CHAPTER1. From Sequence to Structure 1-0. Overview : Protein Function and Architecture Figure 1-1. Four examples of biochemical functions performed by proteins 1-0. Overview : Protein Function and Architecture Figure 1-1. Four examples of biochemical functions performed by proteins 1-0. Overview : Protein Function and Architecture Figure 1-1. Four examples of biochemical functions performed by proteins 1-0. Overview : Protein Function and Architecture Sheet & strand Figure 1-1. Four examples of biochemical functions performed by proteins 1-0. Overview : Protein Function and Architecture Figure 1-2. Levels of protein structure illustrated by the catabolite activator protein 1-1. Amino Acids Figure 1-3. Amino-acid structure and the chemical characters of the amino-acid side chains Structure and Stereoisomerism of a-Amino Acids Ca : a-carbon (chiral) NH3+ : amino group COO- : carboxyl group R : functional group (side chain) Absolute Configuration : S Absolute Configuration : R Left, Counter-Clockwise Right, Clockwise Only L-amino acids are constituents of proteins. 1-1. Amino Acids Figure 1-3. Amino-acid structure and the chemical characters of the amino-acid side chains 1-1. Amino Acids Figure 1-3. Amino-acid structure and the chemical characters of the amino-acid side chains The amino-acid side chains have different tendencies to participate in interactions Hydrophobic residues: van der Waals interactions – tendency to avoid contact with water and pack against each other hydrophobic effect - Ala & Leu are strong helix-favoring residues, Proline are not because its backbone nitrogen isn’t available for H-bond - Aromatic side chain of Phe participates in weakly polar interactions Hydrophilic residues : Hydrogen bonds to one another, to peptide backbone, to polar organic molecules, and to water. - pKa shift: Asp & Glu (57 in hydrophobic interior or nearby (-) charge), Lys (106 in ?) - His: most versatile, most often found in enzyme active sites, pKa is 6, neutral, proton donator and acceptor - Arg: completely protonated at neural, compared to Lys? - Cys: common in enzyme active site, most powerful nucleophile. Compared to Ser? Amphipatic residues : both polar and nonpolar character - Lys: hydrophic charged region, long hydrophobic region (methylene) involved in van der Waals interactions with hydrophobic side chains - Tyr: pKa is 9, in some enzyme active site, hydroxyl group can be donor and acceptor of H-bond. Aromatic ring can form weakly polar interactions - Trp: similar to Tyr, indole amide hydrogen don’t ionize. - Met: least polar among amphipatic residues, thioether sulfur is excellent ligand for metal ions. van der waals interaction - caused by transient dipoles, the momentary random fluctuation in the distribution of the electrons of any atoms Hydrophobic Effects? 1-2. Genes and Proteins The genetic code is degenerate Figure 1-4. The genetic code 1-2. Genes and Proteins Splicing And ? Figure 1-5. The flow of genetic information in prokaryotes (left) and eukaryotes (right) Alternative splicing can lead to truncated proteins, proteins with different stretches in the middle, and frameshifts. Coding sequences can also be modified by RNA editing; some nucleotides can be changed and additional nucleotides inserted into the mRNA sequence before translation. Genetic code organization Single-base changes (single-nucleotide polymorphism) in the third position in a codon produce the same amino acid. Changes elsewhere in the codon produce a different amino acid, but with the same physical-chemical propherties. The second base specifies if the amino acid is polar or hydrophobic. Conservative substitutions. 1-2. Genes and Proteins Figure 1-6. Table of the frequency with which one amino acid is replaced by others in amino-acid sequences of the same protein from different organisms 1-3. The Peptide Bond Figure 1-7. Peptide bond formation and hydrolysis 1-3. The Peptide Bond Resonance of peptide bond - Polarity, Dipole moment - partial double-bond character Figure 1-8. Schematic diagram of an extended polypeptide chain 1-3. The Peptide Bond Ramachandran plot Figure 1-9. Extended polypeptide chain showing the typical backbone bond lengths and angles 1-4. Bonds that Stabilize Folded Proteins Folded proteins are stabilized mainly by weak noncovalent interactions 1 kcal = 4.2 kJ Figure 1-10 Table of the typical chemical interactions that stabilize polypeptides 1-5. Importance and Determinants of Secondary Structure Peptide back bone C=O and N-H tend to hydrogen bond with one another, which result in the secondary structure. Especially in the interior of proteins. Figure 1-11. Ramachandran plot Rotational Properties of Peptide Bonds Peptide bonds are rigid… But, the bonds containing the a-carbon between two peptide bonds can be rotated from -180o to +180o. : the angle of rotation about the bond between the nitrogen and the a-carbon y : the angle of rotation about the a-carbon and the carbonyl carbon 1-5. Importance and Determinants of Secondary Structure Prediction of secondary structure elements from a. a sequence is accurate to only about 70%. convenient way of fold classification N+3 N Figure 1-12.Typical beta turn beta turn, reverse turn, hairpin turn 1-6. Properties of the Alpha Helix N N+4 Figure 1-13.The alpha helix 1-6. Properties of the Alpha Helix Lipid bilayer thickness? 30A. To span the cell membrane, how long helix? at least 20 residues long helix All helices in real protein structures are right-handed. Why? because of steric hindrance caused by L-configuration Helix dipole increase with increasing length of the helix At the N-terminal ends of helices negative side chain Figure 1-14.Table of helical parameters 1-6. Properties of the Alpha Helix Alpha helices can be amphipathic, with one polar and one nonpolar face Figure 1-15.View along the axis of an idealized alpha-helical polypeptide 1-6. Properties of the Alpha Helix 1) Special examples of a-helix Collagen : bone, tendon, ligament and blood vessel Every third residue, glycine (GlyXY)n, X & Y proline -Proline lacks N-H groups, hydroxylation! Figure 1-16.The structure of collagen • Collagen: the most abundant protein of mammals, main fibrous component of skin, bone, tendon, cartilage, and teeth. (피부미용) 2) Special examples of a-helix Coiled-coil protein • Structural support for Cells and Tissues a-keratin: left-handed superhelix of two right-handed a helices. from wool & hair, intermediate filaments in cytoskeleton, muscle protein (myosin & tropomyosin) Heptad repeats; Every seventh residue in each helix, Leu holds two helix by van der Waals interactions disulfide bond crosslinks: fewer – flexible, more – harder (horns, claws etc) 1-7. Properties of the Beta Sheet Figure 1-17.The structure of the beta sheet 1-7. Properties of the Beta Sheet No Plain b-sheet Only twisted b-sheet. Why? Stability & integrity of b-sheet depends on # of b-strands Figure 1-18. Two proteins that form a complex through hydrogen bonding between beta strands (the Rap-Raf complex, PDB 1gua) 1-7. Properties of the Beta Sheet b strands usually have a pronouced right-handed twist, due to steric effects arising from the L-amino acid configuration. Figure 1-19. Beta barrel ; closed cylinder, retinol-binding protein 1-8. Prediction of Secondary Structure A-helices prediction Is easier than b-sheet Figure 1-20.Table of conformational preferences of the amino acids 1-8. Prediction of Secondary Structure Figure 1-21. An example of secondary structure prediction 1-9. Folding The structure of a protein is directly determined by its primary structure Figure 1-22. Folding intermediates 1-9. Folding Competition between self-interactions and interactions with water drives Protein folding Figure 1-23. Highly simplified schematic representation of the folding of a polypeptide chain in water Computational prediction of folding is not yet reliable Ab initio method - Equilibrium conformation is the global free-energy minimum - potential energy parameter is accurate (H-bond, van der Waals etc) - key intermediates? - oligomerization can not be addressed although very many globular proteins are oligomeric. Protein folding funnel The hydrophobic environment of a membrane permits only all-helical and all-beta-barrel integral membrane • The polar amide and carbonyl group should hydrogen bond to one another because water can’t involve in H-bonds 1-10. Tertiary Structure The condensation of multiple secondary structural elements leads to tertiary structure •Two proteins with similar secondary structure elements but different tertiary structures Figure 1-24. Comparison of the structures of triosephosphate isomerase and dihydrofolate reductase 1-10. Tertiary Structure - loops Found at the surface of protein and Exposed to the solvent Sites for protein recognition, ligand Binding and membrane interaction Often mutation sites without changing the core structure Often move as rigid bodies because their side chain pack together Figure 1-25.Variable loops 1-10. Tertiary Structure • Protein crystals contain more than 50% waters in their volumn • hydration shell • a few water inside the protein makes important interactions as part of the tertiary structure Figure 1-26. Porcine pancreatic elastase showing the first hydration shell surrounding the protein 1-10. Tertiary Structure The atoms are packed as closely as in a solid. A few cavities and small channels provide some flexibility. Packing by ionic bonds, H-bonds, and van der Waal interactions. Packing types Figure 1-27. Cut-away view of the interior of a folded protein 1-10. Tertiary Structure The protruding side chains of one helix fit into grooves along the surface of the other helix: ridges and grooves Figure 1-28. Packing motifs of a helical structure 1-11. Membrane Protein Structure H-bond in a completely nonpolar environment are considered stronger than in water Figure 1-29. A segment of a simulated membrane bilayer 1-11. Membrane Protein Structure a helices are the most common secondary structure in membrane proteins Hydrophobic side chain Polar side chains interacting with polar head group of the lipids and each other Figure 1-30. The three-dimensional structure of part of the cytochrome bc1 complex 1-11. Membrane Protein Structure Average hydrophobicity of an eight-residue along the sequence : hydrophathy Prediction of membrane a helices ; 20 consecutive hydrophobic residues Figure 1-31. Hydropathy plot of the Rhizobium meliloti protein DctB 1-11. Membrane Protein Structure • Membrane b sheet prediction is difficult because of various b strands tilt (above 8-9 a.a) • all sheet are antiparallel sheet with short polar turns • all b barrel: hydrophobic side chains in surface, polar side chain inside of the barrel common in channel How about a mixed structure of b sheet and a helix? Figure 1-32. The three-dimensional structure of the all-beta transport protein FhuA 1-11. Membrane Protein Structure •View looking down the channel •The pore-forming loop, 2 K+ ions • Nobel prize in chemistyr 2003 - Roderick Mackinnon (kcsA K+ channel) - Peter Agre (water channel) Figure 1-33.Three-dimensional structure of the bacterial potassium channel The Nobel Prize in Chemistry 2003 "for structural and mechanistic studies of ion channels" Roderick Mackinnon succeeded in determining the first high-resolution structure of an ion channel, the kcsA K+ channel from streptomyces lividans. Water channel and ion channel 13.6 Specific channels increase the permeability of some membranes to water -Some tissues need to transport water. -kidney, secretion of saliva and tears. ※ Aquaporin (Peter Agre in red-blood-cell membrane) -Water channel. 24kda membrane protein -6 membrane spanning helices. -Positive residues in the center prevent the transport of protons through aquaporin; maintain proton gradients Ion channel Potassium and sodium ion For the potassium ions the distance to the oxygen atoms in the ion filter is the same as in water. The sodium ions, which are smaller, do not fit in between the oxygen atoms in the filter. This prevents them from entering the channel. The atomic radius K+ is 1.33 Å and that of Na+ is 0.95 Å . The different kinds of K+ channels gating (opening) • ligand gated - dependent on the intracellular Ca2+ concentration - the level of certain G-protein subunits in the cell - cytoplasmic or extracellular domains for binding ligands. • voltage gated - the membrane voltage-dependent. - integral membrane domains for sensing voltage differences. The Structure of the Potassium Channel four usually identical subunits that encircle with four-fold symmetry → inner, outer, pore α -helix Molecular surface of KcsA and contour of the pore the entire internal pore The ion conduction pore of K+ channels a pore helix (red) and a selectivity filter (gold). Blue mesh shows electron density for K+ ions and water along the pore. The ion conduction pore of K+ channels Selective ion conduction K+ channels conduct K+ ions specifically because the selectivity filter contains multiple binding sites that mimic a hydrated K+ ion’s hydration shell. Potassium channels achieve high conduction rates by exploiting electrostatic repulsion between closely spaced ions and by coupling the conformation of the selectivity filter to ion binding within the filter. 1-12. Protein Stability : Weak Interactions and Flexibility The folded protein is a thermodynamic compromise •Stability is a net loss of free energy (entropy + enthalpy) Free energy difference between the folded and unfolded states; ~21-42kJ/mole, marginally stable. •Folding decrease the entropy of the proteins ; but increase of water entropy is much bigger (hydrophobic effect) Water’s role in weak interactions 1.Small enthalpy difference. 2. Hydrophobic effect – entrophy (nonpolar groups in water tend to be Surrounded by a cluster of water mols) Figure 1-34. Illustration of the ordered arrays of water molecules surrounding exposed hydrophobic residues in bovine pancreatic ribonuclease A 1-12. Protein Stability : Weak Interactions and Flexibility Protein structure can be disrupted by a variety of agents • CD shows protein conformation. • Denaturants (urea, guanidinium hydrochloride, SDS) competes for H-bonds with polar groups of the back bone and side chains. • Features of themophilic proteins - more salt bridges - more hydrophobic interactions & shorter loops Figure 1-35. Computed circular dichroism spectra for the evaluation of protein conformation 1-12. Protein Stability : Weak Interactions and Flexibility The marginal stability of protein tertiary structure allows proteins to be flexible All chemical bonds are flexible because of Vibration and rotation Proteins are much more flexible because of weak interactions to break and reform frequently – Thermal fluctuation is essential for protein function (up to a few angstroms) - Adjust to the binding of ligand or substrate - Water penetration into the interior of the protein Figure 1-36. Results of a molecular dynamics simulation of two interacting alpha helices 1-13. Protein Stability : Post-Translational Modifications Covalent bonds can add stability to tertiary structure 1.Disulfide bond between cysteine side chains -Oxidation of two sulfhydryl groups in ER - not found in most intracellular proteins, but common in secreted proteins Figure 1-37. The structure of the small protein bovine pancreatic trypsin inhibitor, BPTI 1-13. Protein Stability : Post-Translational Modifications 2. Coordinate covalent bonds -Coordination of a metal ion to side chains or water molecules - Ca2+ , Zn2+ most common -removal of the metal ions can leads to denaturation (EDTA) Figure 1-38. Stabilization by coordinate covalent bonds 1-13. Protein Stability : Post-Translational Modifications 3.Organic or organometallic cofactor at the active site Pyridoxal phosphate in D-amino acid aminotransferase Methionine S and heme iron in Cytochrome C Porphyrin cofactor - covalent bonds Heme iron in myoglobin PQQ in polyamine oxidases Figure 1-39. Examples of stabilization by cofactor binding 1-13. Protein Stability : Post-Translational Modifications Glycosylation at serine, threonine or asparagine residues -N-glycosylation site : NxS/T motif -Most important for protein stability, folding, protein-protein recognition (blood cell surface proteins, prevent cells from sticking to one another, cell walls) -Deglycosylation can lead to unfolding or to aggregation. Stability change! -Generally not alter the tertiary structure of a protein, Crystal Structure? Phosphorylation and N-acetylation are reversible and conformational switches Histones or DNA methylases & Demethylases (JMJD2A, LSD1) Histone acetylation & deacetylation (CBP, HDACs) Figure 1-40.Table of post-translation modifications affecting protein stability Each histone is organized in two domains, a characteristic ‘histone fold’ and an unstructured N-terminal ‘tail’. The histone-fold domains constrain the DNA in a central core particle and, thereby, restrict access of DNA-binding proteins. This histone tail is a flexible amino terminus of 11-37 residues. Several positively charged lysine side chains in the histone tail may Interact with linker DNA, and the tails of one nucleosome likely interact with Neighboring nucleosomes higher-order coiling. The histone tail lysine, especially those in H3 and H4, undergo reversible acetylation and deacetylation by enzymes such as CBP (P300) and HDACs In the acetylated form, the positve charge of the lysine e-amino group is neuralized. This eliminate its interaction with a DNA phosphate group. So the greater the acetylation of histone N-terminus, the less likely chromatin is to form condensed 30-nm fibers and possibly higher-order folded structures. Sites of Histone Tail Modifications Epigenetics edited by Allis et al. (2007) 1-14. The Protein Domain Globular proteins are composed of structural domains -Domain is a structural and functional unit composed of generally continuous amino acids (50~200 a.a.) -Domains have hydrophobic cores Tetramerization and DNA-binding domain Lac repressor tetramer binding to DNA Interuption! Alanine racemase 1-14. The Protein Domain Multidomain proteins probably evolved by the fusion of genes that once coded for separate proteins -A single gene is assumed to have been duplicated in tandem -The more ancient the gene duplication, the more time for mutation to happen - examples Two subunits Figure 1-43. Structures of thioesterase and thioester dehydrase 1-14. The Protein Domain Gene duplication within a single structural domain Two nearly identical domains Figure 1-44. Structure of gamma-crystallin, eye-lens protein 1-14. The Protein Domain The number of protein folds is large but limited - Protein folds are used repeatedly in different combinations Figure 1-45. Structures of tryptophan synthase and galactonate dehydratase 1-15. The Universe of Protein Structures Proteins are grouped into families on the basis of the domains, whose functions are classified. Tempting but not the all case. Kinases, a/b hydrolase SH2: phospho-tyr, SH3: proline-rich PH: bind to membrane Figure 1-46. Schematic diagram of the domain arrangement of number of signal transduction proteins 1-15. The Universe of Protein Structures Common TIM barrel of eight-stranded parallel beta barrel But different biochemical functions; exceptional case! -use NADPH to reduce sugars. -hydrolyze phosphate goups Figure 1-47. Structures of aldose reductase (left) and phosphotriesterase (right) Both enzymes catalyze the same reaction but they have no structural Similarity to each other in a.a. sequence and tertiary structure Figure 1-48. Structures of aspartate aminotransferase (left) and D-amino acid aminotransferase (right) The modular structure of protein structure allows for sequence insertions and deletions Q: How long polypeptides domains can be inserted in or deleted from a protein structure without disrupting structure? Q: Insertions and deletions nearly always occur in the surface loop. Why? 1-16. Protein Motifs may be defined by their primary sequence or by the arrangement of 2nd structure elements Protein Motif • sequence motif/structural motif • functional motif Zinc finger motif – sequence motif CXX(XX)CXXXXXXXXXXXXHXXXH Figure 1-49. Zinc finger motif 1-16. Protein Motifs • Functional motif • Structural motif Human growth hormone Figure 50. Helix-turn-helix Four-helix bundle motif 1-16. Protein Motifs •Identifying motifs from sequence is not straightforward. •Functional motifs are detected from the structure rather than the sequence. Figure 1-52. Catalytic triad of serine protease (a) subtilisin, (b) chymotrypsin. 1-17. Alpha Domains and Beta Domains Group domain folds into 5 classes, based on the predominant secondary structure. •Alpha domains: •Beta domains: only beta sheet •Alpha/beta domains: beta strands with connecting helical segments •Alpha+beta domains: separate beta sheet and helical regions •Cross-linked domains: secondary structure are stabilized by disulfide bonds or metal ions 1-17. Two common motifs for alpha domains Four-helix bundle - Common in hormones Oxygen-strage protein in marine worms 4 helices, 20 degree tilt, Myohemerthrin Globin fold Oxygen-strage protein 8 helices, 90 and 50 degree tilt Hydrophobic pocket for organometalic Myoglobin 1-17. beta domains contain strands connected in two distinct ways 1. Connected to adjacent strand; Up-and-down structural motif Neuraminidase beta-propeller domain 2. Connected to 3rd strand; Greek key motif Pre-albumin 1-17. Antiparallel beta sheets can form barrels and sandwiches All-beta domains contain antiparallel beta strcuture, the strands of which are connected with beta turns and larger loops Beta sandwiches -Antiparallel beta sheets pack against each other; one face of b sheet orients to solvent, and the other face orient toward the hydrophoic core. -two greek-key motifs Figure 1-56. Immunoglobulin 1-17. Beta Domains; variation Jelly roll: variation of beta sandwich Fibrous protein silk: two-sheet structure Sequence: GAGSGAGSGAGSG Figure 1-59. Bacteriochlorophyll A protein 1-18. Alpha/Beta, Alpha+Beta and Cross-Linked Domains In alpha/beta domains, each strand of parallel beta sheet is usually connected to the next by an alpha helix giving rise to beta-alpha-beta-alpha units Right-handed crossover connection Right-handed twist of the beta sheet prefered the right-handed crossover topology left-handed crossover connection Figure 1-60. Crossover connection between parallel beta strands 1-18. Alpha/Beta Domains Two major families of a/b domains: a/b barrel and a/b twist •a/b barrel, TIM barrel Triosphosphate isomerase •Parallel & Nonpolar beta strand followed by a amphipathic Alpha helix, repeated eight times Figure 1-61. Alpha/beta domains •a/b twist, Semi-aldehyde dehydrogenase parallel b strands protected from water by a helices coating Nucleotide-binding fold 1-18. Alpha+Beta Domains Alpha+beta domains have independent helical motifs packed against beta sheet; segregated! MHC Figure 1-62. Alpha+beta saddle,TATA-binding protein 1-18. Cross-Linked Domains In small irregular domains, disulfide bridges and metal ions form cross-links Scorpion toxin -Very stable to proteolytic Digestion and heat denaturation -No hydrophobic core but stabilized by 4 disulfide bonds Figure 1-63. Disulfide-linked protein 1-18. Cross-Linked Domains 2 histidines and 2 cysteines Coordinate a zinc ion. Too small to have hydrophobic core The most abundant one in the human Figure 1-64. Zinc finger 1-19. Quaternary Structure : General Principles Oligomers: composed of more than one polypeptide chain subunits Hemoglobin Most common Figure 1-65. Schematic representations of different kinds of oligomers 1-19. All specific intermolecular interactions depend on complementarity Protein surface is irregular. What doest enable proteins to bind specific molecules? Shape complementarity is necessary for large number of weak interactions and to maximize the strength of interactions ((H-bonds and van der Waals) Figure 1-66. “Open-book” view of the complementary structural surfaces that form the interface between interleukin-4 (left) and its receptor (right) 1-19. Shape complementarity For stable complex, Bond strength should be greater than about 15-20 kJ/mole. So shape complementarity is necessary. tropomyosin Figure 1-67. Coiled-coil alpha-helical interactions 1-19. Shape complementarity C C Heptad repeat Transcription factors of leucine zipper On monomers are disordered But fold on dimerization by hydrophobic Interactions N N Figure 1-68. Peptide-peptide interactions in the coiled coil of the leucine zipper family of DNA-binding proteins 1-20. Quaternary Structure : Intermolecular Interfaces Formation of intermolecular interface Is mediated by hydrophobic interactions, Hydrogen bonds and salt briges Including metal-ion ligation and disulfide bond Hemoglobin: several intersubunit salt bridges Depending on pH, alter the relative orientation of the subunits & the affinity for oxygens Very stable oligomers tend to bury a large hydrophobic surface area between subunits while polar interactions more easily break Figure 1-69. Water molecules at a subunit interface 1-20. Quaternary Structure : Intermolecular Interfaces Rap Raf Figure 1-70. Oligomerization by beta sheet formation 1-20. Inappropriate quaternary interactions induce disease Hydrophobic patch from the mutation in b2 subunit (Gln Val) Thick fiber Figure 1-71. Sickle-cell hemoglobin 1-20. Quaternary Structure : Intermolecular Interfaces Oligomeric proteins are more susceptible to disruption by mutation Monomeric proteins; loss of function occurs in homozygous conditions Oligomeric proteins: loss of function may occur in heterozygous conditions Why oligomer in terms of evolution? Figure 1-72. Dominant-negative phenotype resulting from hydrophobic interactions between mutant and normal subunits of a dimeric protein 1-21. Protein assemblies built of identical subunits are usually symmetric Asymmetric complex Figure 1-73.The human growth hormone-receptor complex 1-21. Protein assemblies built of identical subunits are usually symmetric Asymmetric unit is Protomer. Figure 1-75. Interactions underlying different geometric arrangements of subunits 1-21. Quaternary Structure: Geometry Figure 1-74. Examples of quaternary arrangements observed for oligomeric proteins 1-21. Quaternary Structure: Geometry rhinovirus proteasome Figure 1-74. Examples of quaternary arrangements observed for oligomeric proteins 1-22. Proteins are flexible Figure 1-76.Table of protein motions 1-22. Conformational fluctuations in domain structure tend to be local Common ligand-induced conformational change is the lid-like movement of a polypeptide segment to cover a ligand-binding site. Figure 1-77.Triosephosphate isomerase 1-22. Protein Flexibility Alternate between distinct conformations Free-energy barrier Figure 1-79.T4 lysozyme 1-22. Protein Flexibility Driving force is provided by ligand-protein Interactions Induced fit Figure 1-80. Aspartate aminotransferase, open and closed forms