Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Designer baby wikipedia , lookup
History of genetic engineering wikipedia , lookup
RNA interference wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Epitranscriptome wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Point mutation wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Primary transcript wikipedia , lookup
Gene expression profiling wikipedia , lookup
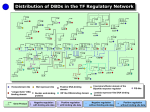
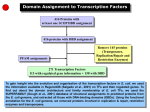
Supplemental methods Computational Kinase Enrichment Analysis Differentially expressed (DE) genes from the three microarray datasets were used individually to identify upstream transcription factors (TFs) with ChIP Enrichment Analysis (ChEA).1 ChEA uses a database of experimentally validated ChIP-chip, ChIPseq and ChIP-PET sites for TFs to examine the enrichment of TF binding sites at a given set of genes as compared to the whole genome. The enriched TFs were ranked by pvalue, and the top 10 TFs from each dataset were considered for further analysis. Seven TFs (HNF4A, PPARG, MITF, CREM, FLI1, KDM5B and RUNX1) were among the top 10 in at least two datasets, and these were further analyzed with Kinase Enrichment Analysis (KEA) to identify the protein kinases that regulate these seven TFs.2 Expression2Kinase (X2K) analysis was subsequently applied to identify the protein kinases that regulate gene expression.3 As before, the top 10 TFs that regulate the DE genes were identified in each dataset and then Genes2Networks4 was used to link these 10 TFs in each dataset to their regulatory networks by known protein-protein interactions.5 The Human Protein Reference Database6 was used to generate the networks. The identified networks contained an average of 126 nodes (proteins) in each of the three datasets. We used these nodes to identify the upstream protein kinases using KEA in each dataset. The common kinases in all the three sets were considered as common upstream kinases in liver tumorigenesis. To characterize the connections among these kinases, they were analyzed through the use of Ingenuity Pathway Analyses (IPA) (Ingenuity Systems). CREM knock down in Human HCC cell lines. Dicer substrate siRNA (DsiRNA) targeting human CREM gene were designed and synthesized by IDT (Integrated DNA Technologies).7 For Knock down assay. DsiCREMs (10pM) were transfected into Huh7 and HepG2 cells with Lipofectamine 2000 (Invitrogen). 48h later, transfected cell were changed to serum free medium to starve overnight. Next day, the cells were subjected to insulin (10nM) stimulation or RNA isolation. The sequence of the DsiCREM is as follow: DsiCREM-1(KD#1): Sequence Strand 5' rCrGrArGrArGrCrUrGrArGrGrCrUrArArUrGrArArArArACA 3' + 5' rUrGrUrUrUrUrUrCrArUrUrArGrCrCrUrCrArGrCrUrCrUrCrGrUrU 3' DsiCREM-2(KD#2): Sequence Strand 5' rCrUrGrGrUrGrArCrArUrGrCrCrArArCrUrUrArCrCrArGAT 3' + 5' rArUrCrUrGrGrUrArArGrUrUrGrGrCrArUrGrUrCrArCrCrArGrUrG 3' - Supplemental figure legends Sfig 1. CAMK2γ is regulated by bile acids during acute liver injury. A. An ingenuity pathway analysis of kinases identified by ChEA and KEA. B-D. Protein regulation network generated from Top 10 Transcription factor of the three HCC dataset: DEN vs control (B), FXR-/- vs WT mice (C) and Human HCC samples vs non-tumor tissue (D). The yellow nodes in the figures represents the Top 10 TF in each dataset while the green nodes represents the Top ranked Kinases. E. Phosphorylation of CAMK2γ at T287 in the liver of the mice treated by 100 mg/kg DEN i.p. injection. Sfig 2. Deletion of CAMK2 γ promotes cell deaths, inflammation, and compensatory hepatocyte proliferation. A. Representative images of H&E and TUNEL staining of the wild-type and CAMK2γ-/- mouse livers corresponding to the mice in Figure 2C. B. Quantification of necrosis and apoptosis in the liver. C. Representative BrdU staining and quantification. Sfig 3. Deletion of CAMK2γ promotes synthesis of inflammatory cytokines. Realtime PCR analyses of the mRNA levels of the hepatic inflammatory cytokines in the wildtype and CAMK2γ-/- mice with DEN and TCPOBOP-induced hepatocellular carcinomas. Sfig 4. Deletion of CAMK2 γ enhances STAT3 activation and decrease p53 proteins. A. Western blotting analysis of the liver lysates from the wild-type and the CAMK2γ -/- mice with DEN/TCPOBOP-induced liver cancer. B. mRNA levels of p53 target genes. C. mRNA levels of STAT3 target genes. Sfig 5. CAMK2γ inhibition promotes AKT activation in liver cells. A. Livers of wildtype and CAMK2γ-/- mice without any treatment. B. Huh7 cells were starved overnight and then pretreated with combination of DMSO, PD98059, rapamycin or KN93. The pretreated cells were then stimulated with 10nM insulin for 30 min. Cell lysate were subjected for western blotting analysis. Sfig 6. IL-6 mRNA expression change after DEN injection in WT and KO mice. Sfig 7. CREM knockdown do not affect Akt activation. A. Scheme of interaction between Top 7 TF and Top 10 Kinase in the KEA analysis. B. Knock down efficiency of CREM in Huh7 and HepG2 cell lines. C. Cell viability test of Huh7 and HepG2 cells after CREM knock down. D. Knocking down CREM do not affect Akt activation. E. Knocking down CREM do not rescue CAMK2γ overexpression induced Akt inhibition. STable 1. Top TFs with enriched targets in each HCC datasets reported by ChEA. P-values of enrichment for input versus database calculated using Fisher Exact Test. STable 2. Top 10 Kinases with enriched from the Top TFs. References 1. Lachmann A, Xu H, Krishnan J, et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010;26:2438-44. 2. Lachmann A, Ma'ayan A. KEA: kinase enrichment analysis. Bioinformatics 2009;25:684-6. 3. Chen EY, Xu H, Gordonov S, et al. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2012;28:105-11. 4. Berger SI, Posner JM, Ma'ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. Bmc Bioinformatics 2007;8. 5. Berger SI, Posner JM, Ma'ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics 2007;8:372. 6. Prasad TSK, Goel R, Kandasamy K, et al. Human Protein Reference Database2009 update. Nucleic Acids Research 2009;37:D767-D772. 7. Kim DH, Behlke MA, Rose SD, et al. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 2005;23:222-6.