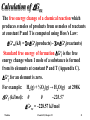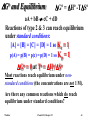Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Temperature wikipedia , lookup
Eigenstate thermalization hypothesis wikipedia , lookup
Chemical potential wikipedia , lookup
Detailed balance wikipedia , lookup
Rate equation wikipedia , lookup
Heat transfer physics wikipedia , lookup
Electrochemistry wikipedia , lookup
Vapor–liquid equilibrium wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Reaction progress kinetic analysis wikipedia , lookup
Determination of equilibrium constants wikipedia , lookup
Gibbs paradox wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Work (thermodynamics) wikipedia , lookup
Marcus theory wikipedia , lookup
Maximum entropy thermodynamics wikipedia , lookup
Enzyme catalysis wikipedia , lookup
George S. Hammond wikipedia , lookup
Thermodynamics wikipedia , lookup
Equilibrium chemistry wikipedia , lookup
Chemical equilibrium wikipedia , lookup
Chemical
Thermodynamics
Chapter 19
A continuation of Chapter 5
Watkins
Chem 1422, Chapter 19
1
Review Chapter 5
1st Law of Thermodynamics
DEuniv = 0
Euniv = Esys + Esurr
DEsys = q + w
w = –PDV (expansion work)
For DV = 0, w = 0
qv = DEsys
For DP = 0, DEsys = qp – PDV
qp = DEsys + PDV = DHsys
Watkins
Chem 1422, Chapter 19
DE and DH are
state functions
q and w are not
state functions
2
Spontaneous Processes
Some reactions occur spontaneously without outside
intervention. For example:
H2O(s, 25 oC) → H2O(l, 25 oC)
ice always melts at room temperature
The reverse of a spontaneous reaction is never
spontaneous:
H2O(l, 25oC) → H2O(s, 25oC)
water never freezes at room temperature
Thermodynamics tries to answer the question:
Under what circumstances is a reaction spontaneous?
Watkins
Chem 1422, Chapter 19
3
Spontaneous Processes
Every system tends spontaneously
toward its lowest energy
Watkins
1
spontaneous
1
2
Dge < 0
2
Gravitational Energy
Gravitational Energy
A ball rolls down a hill spontaneously because its gravitational energy
at the bottom is less than its gravitational energy at the top: Dge < 0
A ball never rolls uphill all by itself; it has to be given extra energy
Dge to climb up the (gravity) hill.
Chem 1422, Chapter 19
2
non-spontaneous
2
1
D ge > 0
1
4
Spontaneous Processes
Every system tends spontaneously
toward its lowest energy
Watkins
R
spontaneous
R
P
Dce < 0
P
Chemical Energy
Chemical Energy
A chemical reaction rolls down a chemical energy hill spontaneously
if the chemical energy of the products is lower than the chemical
energy of the reactants: Dce < 0
A non-spontaneous reaction can be forced to proceed by giving it
extra energy Dce.
P
non-spontaneous
P
R
Dce > 0
Chem 1422, Chapter 19
R
5
Thermodynamically Reversible Reactions
Chemical Energy
A chemical reaction at equilibrium is on a flat chemical
energy surface (Dce = 0). A system at equilibrium is not
spontaneous; in thermodynamics, a system at
equilibrium is called reversible.
Watkins
R
equilibrium
P
R
P
Dce = 0
Chem 1422, Chapter 19
6
ThermodynamicallyReversible Reactions
A reversible reaction goes forward or backward just by
adding or subtracting heat:
H2O(l, 1 atm, 273K) ⇌ H2O(s, 1 atm, 273K)
To freeze 1 mol of water to form 1 mol of ice,
-q = -DHfus of heat is removed from the system.
To reverse the process, +q = +DHfus of heat must be
added to 1 mol of ice to form 1 mol of water.
Therefore, converting between 1 mol of ice and 1 mol
of water at this T and P is a reversible process.
Watkins
Chem 1422, Chapter 19
7
Reversible and Irreversible Reactions
A reaction at equilibrium (Q = K) is reversible and
therefore non-spontaneous.
A reaction which is not at equilibrium (Q K) is
irreversible;
if Q < K, the reaction is spontaneous in the forward
direction;
if Q > K, the reaction is spontaneous in the reverse
direction.
Watkins
Chem 1422, Chapter 19
8
Spontaneous Reactions
Watkins
R
spontaneous
R
P
Dce < 0
P
Chemical Energy
Chemical Energy
We predict the spontaneity and direction of a chemical
reaction by comparing the values of Q and K;
thermodynamics cannot predict the speed at which the
reaction will occur (that's kinetics!).
So what is “Chemical Energy”? The first thing to
consider is DH.
P
non-spontaneous
P
R
Dce > 0
Chem 1422, Chapter 19
R
9
Spontaneous Reactions
We predict the spontaneity and direction of a chemical
reaction by comparing the values of Q and K;
thermodynamics cannot predict the speed at which the
reaction will occur (that's kinetics!).
So what is “Chemical Energy”? The first thing to
consider is DH.
Many spontaneous reactions are exothermic; they seem
to roll down an enthalpy hill. (Dce = DH?)
But some spontaneous reactions are endothermic, so
Dce DH. But is Dce = DE?
There are some spontaneous processes for which there is
no apparent energy change at all!
Watkins
Chem 1422, Chapter 19
10
Spontaneous Processes
Expansion of an Ideal Gas
Initial state: two 1 L flasks are connected by a closed
stopcock; one flask is evacuated and the other
contains 1 atm of an ideal gas.
Watkins
Chem 1422, Chapter 19
11
Spontaneous Processes
Expansion of an Ideal Gas
Process: when the stopcock is opened, the gas
spontaneously expands to fill both flasks.
Watkins
Chem 1422, Chapter 19
12
Spontaneous Processes
Expansion of an Ideal Gas
Final state: the two flasks are connected by an open
stopcock; each flask contains gas at 0.5 atm.
Watkins
Chem 1422, Chapter 19
13
Spontaneous Processes
Expansion of an Ideal Gas
The expansion of an ideal gas is isothermal (DT = 0) so
no heat is transferred (q = 0; adiabatic).
The flasks are rigid so there is no expansion work
(w = 0).
Thus, DE = q + w = 0
Watkins
Chem 1422, Chapter 19
14
Spontaneous Processes
Expansion of an Ideal Gas
If DE = 0 and DH = 0, why does the gas expand
spontaneously (Dce < 0)?
The spontaneous expansion of an ideal gas rolls down a
chemical energy hill created by ENTROPY
Watkins
Chem 1422, Chapter 19
15
Entropy - S
Entropy is a measure of the probability and disorder of a
system.
Analogy: unwrap a new deck of cards; it is arranged in
suits and is highly ordered – the new deck has low
entropy. Now throw the deck into the air...
the cards will probably land in a disordered jumble:
high S.
the cards will (probably) never land in a neat stack
with the cards in suits: low S.
high S = high probability = disorder
low S = low probability = order
Watkins
Chem 1422, Chapter 19
16
Entropy - S
high S = high probability = disorder
low S = low probability = order
Example: adiabatic expansion of a gas...
Gas molecules are in constant and random motion. In a
small volume, the molecules have less room for their
random motion, but in a larger volume, the molecules can
move in a more random (disordered) way. Thus...
Gas in a smaller volume
low S & low probability
Watkins
DS > 0
Gas in a larger volume
high S & high probability
Chem 1422, Chapter 19
17
Entropy - S
high S = high probability = disorder
low S = low probability = order
Example: ice has low S because the molecules are held in
ordered positions by H-bonds.
Ice spontaneously melts at room temperature
because liquid water is composed of molecules in
random motion (high S).
H2O(s,298K) → H2O(l,298K)
DS > 0
Solid = low S
Liquid = higher S
Gas = highest S
Watkins
Chem 1422, Chapter 19
18
Entropy - S
high S = high probability = disorder
low S = low probability = order
Example: an ionic solid
dissolves in water:
the ions leave the crystal
and float off into the
solution (increasing S)
the water organizes into
hydrates about the ions
(decreasing S)
DSdiss, the total entropy of
dissolution is the sum
(usually > 0)
Watkins
Chem 1422, Chapter 19
19
Measuring DS
The entropy of a system is a state function:
DSsys = Sfinal – Sinitial
It can be measured in a reversible system.
For water and ice in equilibrium at 0 oC, the two states
can be reversibly exchanged by adding or subtracting
heat (DHfus = qrev = 6010 J/mol) at constant T = 273K:
qrev
DHfus
DSrev = T = T
ice → water @ 273K, qrev > 0: DSrev = +22.0 J/mol-K
water → ice @ 273K, qrev < 0: DSrev = -22.0 J/mol-K
Watkins
Chem 1422, Chapter 19
20
Measuring DS
Any chemical reaction, if carried out in a closed system,
will come to equilibrium at the T and P specified by
the experimenter:
LHR(states,T,P) ⇌ RHR(states,T,P)
Furthermore, every reaction has a heat of reaction at
temperature T, DHrx,T , which can be calculated using
Hess’s Law and a table of Heats of Formation.
Therefore the entropy change for the reversible reaction
at temperature T, DSrx,T , can be computed:
DHrx,T
DSrev = DSrx,T =
T
Note that only values at T = 298K can be computed
from the data in Appendix C.
Watkins
Chem 1422, Chapter 19
21
Measuring DS
Now consider an irreversible (spontaneous) reaction
(e.g., a mixture of water and ice at room temperature).
It has been found experimentally that for all spontaneous
(non-equilibrium) reactions:
DSirrev > DSrev
The concept of entropy, and the rules governing its
behavior, have been formulated as the
2nd Law of Thermodynamics
Watkins
Chem 1422, Chapter 19
22
Second Law of Thermodynamics
DSsys may be + or –, but DSuniv is always positive!
The change in entropy of the universe is the sum of the
change in entropy of the system and the change in
entropy of the surroundings
DSuniv = DSsys + DSsurr
For every reversible (equilibrium) process
DSsys = -DSsurr
DSuniv = 0
For every irreversible (spontaneous ) process
DSsys > -DSsurr
DSuniv > 0
Watkins
Chem 1422, Chapter 19
23
Second Law of Thermodynamics
DSsys may be + or –, but DSuniv is always positive!
What does this mean?
TDS is the energy associated with entropy; TDS is waste
energy which cannot be used for any purpose!
But the total amount of energy in the universe is constant
(First Law)!
Every spontaneous process that occurs at T > 0 converts
some of the energy of the universe into waste energy.
Once all of the energy in the universe is converted to waste
energy, there can be no more spontaneous processes.
The universe is running down!
Watkins
Chem 1422, Chapter 19
24
Molecular Interpretation of Entropy
Absolute Entropy of State
high S
g
l
low S
s
Adding heat increases
both entropy and
temperature ...
except at a phase
change, where added
heat increases only
entropy.
The entropy of a
perfect crystal at 0 K
is 0 (3rd Law of
Thermodynamics)
Watkins
Chem 1422, Chapter 19
25
Molecular Interpretation of Entropy
Entropy Change vs. Amount of Gas
Any reaction that increases the number of gas
molecules leads to a increase in entropy
2NO2(g)
→ 2NO(g) + O2(g)
2 moles gas
lower S
3 moles gas
higher S
DS > 0
Entropy Change vs. Volume of Gas
Entropy increases as a gas expands
Watkins
Chem 1422, Chapter 19
26
Spontaneous Reactions
Watkins
R
spontaneous
R
P
Dce < 0
P
Chemical Energy
Chemical Energy
A spontaneous (non-equilibrium) reaction always
rolls down a “chemical energy” hill which clearly
must involve entropy.
So what is “Chemical Energy”?
We are now ready to answer this question.
P
non-spontaneous
P
R
Dce > 0
Chem 1422, Chapter 19
R
27
Spontaneous Reactions
For DP = 0, the total heat energy (DH) is divided
into useful heat energy (DG) and waste heat
energy (TDS):
DH = DG + TDS
DG = DH - TDS
G is Gibbs Free Energy
G is free to cause spontaneous reactions
G is the chemical energy that drives a
spontaneous chemical reaction.
Watkins
Chem 1422, Chapter 19
28
Thermodynamic Energies
aA + bB ⇌ cC + dD
EA
HA
SA
GA
EB
HB
SB
GB
EC
HC
SC
GC
ED
HD
SD
GD
DErx
DHrx
DSrx
DGrx
Only these three are
used in this course
(see Appendix C)
Each reagent in a reversible reaction contains four specific
molar energy quantities called
total internal energy E kJ/mol
“heat content” or enthalpy H kJ/mol
entropy S J/mol.K (the energy quantity is TS J/mol)
Gibbs free energy G kJ/mol
The energy changes for the reaction are (Hess’s Law)
DErx = cEC + dED – aEA – bEB
Watkins
Chem 1422, Chapter 19
29
Calculation of DHrx
The heat of a chemical reaction which produces n moles
of products from m moles of reactants at constant P and
T is computed using Hess's Law:
DHorx(kJ) = SnDHof(products) - SmDHof(reactants)
Standard heat of formation DHof is the enthalpy change
when 1 mole of a substance is formed from its elements
at constant P and T (Appendix C).
DHof for an element is zero.
For example:
DHof (kJ/mol):
H2(g) + ½O2(g) → H2O(g) at 298K
0
0
-241.82
DHorx = –241.82 kJ/mol
Watkins
Chem 1422, Chapter 19
30
Calculation of DGrx
The free energy change of a chemical reaction which
produces n moles of products from m moles of reactants
at constant P and T is computed using Hess's Law:
DGorx(kJ) = SnDGof(products) - SmDGof(reactants)
Standard free energy of formation DGof is the free
energy change when 1 mole of a substance is formed
from its elements at constant P and T (Appendix C).
DGof for an element is zero.
For example:
DGof (kJ/mol):
H2(g) + ½O2(g) → H2O(g)
0
0
at 298K
–228.57
DGorx = –228.57 kJ/mol
Watkins
Chem 1422, Chapter 19
31
Calculation of DSrx
The entropy change for a chemical reaction which
produces n moles of products from m moles of reactants
at constant P and T is computed using Hess's Law:
DSoT(J/K) = SnSoT(products) - SmSoT(reactants)
Standard molar entropy SoT is the absolute entropy of
1 mole of a substance in its standard state at constant P
and T (Appendix C, units J/mol.K)
SoT for an element is not zero.
H2(g) + ½O2(g) → H2O(g)
For example:
So298 (J/mol.K)
130.6
205.0
at 298K
188.8
DSorx = –44.3 J/K
Watkins
Chem 1422, Chapter 19
32
DG = DH - TDS
Gibbs Free Energy
G is the chemical energy that drives a reaction.
RHR (G ) DG = G
– Glhr < 0
The forward rxn is driven harder than the reverse rxn; the
forward rxn is spontaneous.
LHR (Glhr)
LHR (G )
lhr
rhr
rhr
RHR (Grhr) DG = Grhr – G > 0
lhr
The reverse rxn is driven harder than the forward rxn; the
reverse rxn is spontaneous.
LHR (Glhr)
RHR (Grhr) DG = Grhr – Glhr = 0
The forward and reverse reactions are driven equally, so
neither rxn is spontaneous: equilibrium!
Watkins
Chem 1422, Chapter 19
33
DGof
Standard Free Energies of Formation, DGf
Subscript f: specifies a formation reaction:
Elements are the only reactants,
One mole of a pure compound is the only
product:
1/ N (g) + 3/ H (g) → NH (g)
2 2
2 2
3
Superscript o: Standard states:
Every solid or liquid is pure (except aq);
Every gas is at 1 atm (partial) pressure
All dissolved substances are 1 M
Watkins
Chem 1422, Chapter 19
34
DGof
Standard Free Energies of Formation, DGf
Temperature is not part of the standard state
specification.
DGf values at 298K are tabulated in App. C
for many substances; these values are
different at other temperatures.
Note that for an element DGof = 0 at all T.
DGof {O2(g)} = 0 kJ/mol at any T
DGof {O(g)} = 230 kJ/mol at 298K
Watkins
Chem 1422, Chapter 19
35
Temperature, DGo, DHo, DSo
For any reaction under standard conditions and at any
temperature
DGorx,T = DHorx,T – TDSorx,T
The only values we can calculate from App. C are for T
= 298K: DGorx,298, DHorx,298 and DSorx,298.
But it has been found that DHorx and DSorx are almost
constant for temperatures between about 200K and
400K, and only change slowly outside this range.
Therefore, we will approximate, at all temperatures:
DHoT ≈ DHo298
DSoT ≈ DSo298
Watkins
Chem 1422, Chapter 19
36
Temperature, DGo, DHo, DSo
Let's rearrange the defining equation:
DGorx,T = DHorx,T - TDSorx,T
DGorx,T = (-DSorx,T)T + DHorx,T
y
=
m
x + b
(a stright line)
x =T: the independent variable
y =DGorx,T: the dependent variable
m = -DSorx,T -DSorx,298: the slope
b =DHorx,T DHorx,298: the intercept
Watkins
Chem 1422, Chapter 19
37
Temperature, DGo, DHo, DSo
There are four kinds of reactions
DGoT = (-DSo298)T + DHo298
4. (DHo > 0) (DSo < 0)
3. (DHo > 0) (DSo > 0)
(+)
4
Non-Spontaneous
2
DGo 0
(-)
(DHo
(DSo
2.
< 0)
< 0)
1. (DHo < 0) (DSo > 0)
Watkins
T
Spontaneous
3
1
Chem 1422, Chapter 19
38
Temperature, DGo, DHo, DSo
1. Exothermic
(DHo < 0) with
increasing entropy
(DSo > 0)
2O3(g) → 3O2(g)
spontaneous at all T
O3 is "unstable" with
respect to O2 at all
temperatures.
(but the reaction is
kinetically slow)
DGoT = (-DSo298)T + DHo298
4
Non-Spontaneous
2
T
Spontaneous
3
1
Watkins
Chem 1422, Chapter 19
39
Temperature, DGo, DHo, DSo
2. Exothermic
(DHo < 0) with
decreasing entropy
(DSo < 0)
DGoT = (-DSo298)T + DHo298
CaO(s) + CO2(g) →
CaCO3(s)
Non-Spontaneous
spontaneous at T < To
nonspontaneous at
T > To
To is the temperature at
which equilibrium is
established under
standard conditions
To = DHo /DSo
Watkins
4
2
T
To
Spontaneous
3
1
Chem 1422, Chapter 19
40
Temperature, DGo, DHo, DSo
3. Endothermic
(DHo > 0) with
increasing entropy
(DSo > 0)
DGoT = (-DSo298)T + DHo298
CaCO3(s) →
CaO(s) + CO2(g)
Non-Spontaneous
spontaneous at T > To
nonspontaneous at
T < To
To = DHo/DSo
4
2
T
To
Spontaneous
3
1
Watkins
Chem 1422, Chapter 19
41
Temperature, DGo, DHo, DSo
4. Endothermic
(DHo > 0) with
decreasing entropy
(DSo < 0)
DGoT = (-DSo298)T + DHo298
3O2(g) → 2O3(g)
Non-Spontaneous
nonspontaneous at
all T
(this reaction has to be
“pushed uphill” by
the input of energy
– UV photons from
the sun)
Watkins
4
2
T
Spontaneous
3
1
Chem 1422, Chapter 19
42
DGo and Equilibrium
Equilibrium at To under
standard conditions
(DGo = 0):
2. Exothermic
(DHo < 0) with
decreasing entropy
(DSo < 0)
or
3. Endothermic
(DHo > 0) with
increasing entropy
(DSo > 0)
To = DHo /DSo
DGoT = (-DSo298)T + DHo298
4
Non-Spontaneous
2
T
To
Spontaneous
3
1
Watkins
Chem 1422, Chapter 19
43
DGo and Equilibrium
DGo = DHo -TDSo
aA + bB ⇌ cC + dD
Reactions of type 2 & 3 can reach equilibrium
under standard conditions:
[A] = [B] = [C] = [D] = 1 so Kc = 1
p(A) = p(B) = p(c) = p(D) = 1 so Kp
=1
DGo = 0 at To = DHo/DSo
Most reactions reach equilibrium under nonstandard conditions (the concentrations are not 1 M).
Are there any common reactions which do reach
equilibrium under standard conditions?
Watkins
Chem 1422, Chapter 19
44
DGo and Equilibrium
DGo = DHo -TDSo
Are there any common reactions that do reach
equilibrium under standard conditions?
Yes! Normal Phase Changes.
Melting (fusion): a pure solid is in equilibrium with its
pure liquid under 1 atm pressure at temperature To
(the normal melting point) :
A(s) ⇌ A(l)
K = 1 (DHofus > 0, DSofus > 0)
DG
DGofus,T = DHofus,T – TDSofus,T
T
To = DHofus,T/DSofus,T ≈ DHofus,298/DSofus,298
0
o
o
when T < T , DG fus,T > 0 (liquid freezes)
when T > To, DGofus,T < 0 (solid melts)
when T = To, DGofus,T = 0, (no spontaneous reaction)
o
o
Watkins
Chem 1422, Chapter 19
45
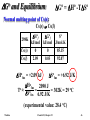
T
DGo and Equilibrium
DGo = DHo -TDSo
Normal melting point of Cs(s):
Cs(s) ⇌ Cs(l)
DHof
298K
kJ/mol
Cs(s)
0
Cs(l)
2.09
DHofus = +2.09 kJ
DGof
kJ/mol
0
0.03
So
J/mol.K
85.15
92.07
DSofus = +6.92 J/K
o
DH
2090 J
To ≈ DSo fus =
= 302K = 29 oC
6.92 J/K
fus
(experimental value: 28.4 oC)
Watkins
Chem 1422, Chapter 19
46
DGo and Equilibrium
DGo = DHo -TDSo
Are there any common reactions that do reach
equilibrium under standard conditions?
Yes! Normal Phase Changes.
Boiling (vaporization): a pure liquid is in equilibrium
with its pure vapor at 1 atm pressure at temperature To
(the normal boiling point) :
A(l) ⇌ A(g) K = 1 (DHovap > 0, DSovap > 0)
DG
DGovap,T = DHovap,T – TDSovap,T
T
To = DHovap,T/DSovap,T ≈ DHovap,298/DSovap,298
0
o
o
when T < T , DG vap,T > 0 (vapor condenses)
when T > To, DGovap,T < 0 (liquid vaporizes)
when T = To, DGovap,T = 0, (no spontaneous reaction)
o
o
Watkins
Chem 1422, Chapter 19
47
T
DGo and Equilibrium
DGo = DHo -TDSo
Normal boiling point of methanol:
CH3OH(l) ⇌ CH3OH(g)
298K
CH3OH(l)
CH3OH(g)
DHof
kJ/mol
-238.6
-201.2
DHovap = +37.4 kJ
DGof
kJ/mol
-166.2
-161.9
So
J/mol.K
126.8
237.6
DSovap = +110.8 J/K
o
DH
37.4 kJ
vap
o
T ≈ DSo =
= 337K = 64 oC
0.111 kJ/K
vap
(experimental value: 64.6 oC)
Watkins
Chem 1422, Chapter 19
48
DGrx,T
DGof,298 is for a formation reaction at 298 K under
standard conditions (appendix C)
DGorx,298 is for any reaction at 298 K under
standard conditions (Hess’s Law + appendix C)
DGorx,T is for any reaction at any temperature
under standard conditions
DGorx,T = DHoT - TDSoT ≈ DHo298 – TDSo298
DGrx,T is for any reaction at any temperature
under any conditions.
Watkins
Chem 1422, Chapter 19
49
DGrx,T
aA + bB ⇌ cC + dD
DGrx,T is the driving force for this reaction under
the specified conditions of T, P, molarities, etc.:
DGrx,T = DGorx,T + RT ln Q
Q specifies concentrations and/or gas pressures
before, during or after the reaction.
To match the units of G (kJ), the required value
of R is 0.008314 kJ/mol.K
DGrx,T = DHorx,T - TDSorx,T + RT ln Q
DGrx,T DHorx,298 - TDSorx,298 + RT ln Q
Watkins
Chem 1422, Chapter 19
50
DGrx,T
aA + bB ⇌ cC + dD
DGrx,T 0: the reaction is driven forward (< 0) or
backward (> 0) spontaneously. In a closed
system, as the reaction proceeds, DGrx,T
approaches zero. In an open system,
reactants and/or products are lost and
DGrx,T never reaches zero.
DGrx,T = 0: the reaction is at equilibrium (only in a
closed system).
Watkins
Chem 1422, Chapter 19
51
Equilibrium and Thermodynamics
C D
Q
a
b
A B
c
aA + bB
cC + dD
Initially [A]i, [B]i, [C]i, [D]i
DGi,T = DGoT + RT ln Qi
d
If DGi,T < 0, reaction proceeds to
the right to reach equilibrium.
As reaction proceeds ...
Q increases and
Qi → Kc
DG increases until ...
DGi → 0
DG = 0 and Qeq = Kc
forward reaction is
spontaneous,
reverse reaction is
non-spontaneous
Watkins
Chem 1422, Chapter 19
52
Equilibrium and Thermodynamics
C D
Q
a
b
A B
c
aA + bB
cC + dD
Initially [A]i, [B]i, [C]i, [D]i
DGi,T = DGoT + RT ln Qi
d
If DGi > 0, reaction proceeds to
the left to reach equilibrium.
As reaction proceeds ...
Q decreases and
Qi → Kc
DG decreases until ...
DGi → 0
DG = 0 and Qeq = Kc
forward reaction is
non-spontaneous,
reverse reaction is
spontaneous
Watkins
Chem 1422, Chapter 19
53
Equilibrium and Thermodynamics
C D
Q
a
b
A B
c
aA + bB
cC + dD
When DG = 0, Q ≡ Kc
Under standard conditions:
Only two kinds of reaction
can reach equilibrium!
DG
And at equilibrium:
d
o
K=1
T = DHo/DSo
Watkins
Chem 1422, Chapter 19
54
Equilibrium and Thermodynamics
C D
Q
a
b
A B
c
aA + bB
cC + dD
When DG = 0, Q ≡ Kc
d
Under non-standard conditions:
DGT = DGoT + RT ln Q
DHo298 - TDSo298 + RT ln Q
Any forward reaction is spontaneous
(DGT < 0) if Q is small enough:
lim Q → 0 => lim lnQ → -∞
Watkins
Chem 1422, Chapter 19
55
Equilibrium and Thermodynamics
C D
Q
a
b
A B
c
aA + bB
cC + dD
When DG = 0, Q ≡ Kc
d
Under non-standard conditions:
DGT = DGoT + RT ln Q
At equilibrium, DGT = 0 and Q = KT
0 = DGoT + RT ln KT
o /RT
-DG
T
KT = e
Watkins
Chem 1422, Chapter 19
56
2SO2(g) + O2(g) ⇌ 2SO3(g)
Calculate DHo298, DSo298, and DGo298 for this reaction.
Standard conditions:
all gases at 1 atm
Gas
298K
SO2
DHof
kJ/mol
-296.9
DGof
kJ/mol
-300.4
So
J/mol.K
248.5
O2
SO3
0
-395.2
0
-370.4
205.0
256.2
Using Hess's Law:
DHo298 = [2(-395.2)] – [2(-296.9)+(0)] = -196.6 kJ
DGo298 = [2(-370.4)] – [2(-300.4)+(0)] = -140.0 kJ
DSo298 = [2(+256.2)] – [2(+248.5)+(+205.0)] = -189.6 J/K
Watkins
Chem 1422, Chapter 19
57
2SO2(g) + O2(g) ⇌ 2SO3(g)
Calculate DGo298 from its defining equation.
DGo298 = DHo298 – TDSo298
DHo298 = -196.6 kJ
DGo298 = -140.0 kJ
DSo298 = -189.6 J/K
DGo298 = (-196.6 kJ)-(298K)(-0.1896 kJ/K) = -140.1 kJ
Match energy units!
Watkins
Chem 1422, Chapter 19
58
2SO2(g) + O2(g) ⇌ 2SO3(g)
Plot DGo as a function of temperature, and find the
equilibrium temperature.
DHo298 = -196.6 kJ
o
o
o
o
DG
DG T = DH T – TDS T
298 = -140.0 kJ
DSo298 = -189.6 J/K
DGoT ≈ DHo298 – TDSo298
Intercept (T = 0): -197 kJ
Slope: +0.19 kJ/K
The equilibrium temperature
at which DGoT = 0 and KT = 1
DGoT = 0 DHo298 – TDSo298
T DHo298/DSo298
= (-196.6 kJ)/(-0.1896 kJ/K)
= 1037 K = 764 oC
Watkins
Chem 1422, Chapter 19
59
2SO2(g) + O2(g) ⇌ 2SO3(g)
Calculate the equilibrium constant at 298K and 350K
o
-DG
298/RT
DHo298 = -196.6 kJ
K298 = e
o
140.0/(0.008314×298)
DG
298 = -140.0 kJ
K298 = e
DSo298 = -189.6 J/K
24
K298 = 3.47×10
K350
R = 8.314 J/mol.K
R = 0.008314 kJ/mol.K
o
-DG
350/RT
=e
DGo350 = DHo350 – (350)DSo350
DHo298 – (350)DSo298
(-196.6 kJ) - (350 K)(-0.1896 kJ/K)
= -130.2 kJ
K350 = e 130.2/(0.008314×350) = 2.74×1019
Watkins
Chem 1422, Chapter 19
60
2SO2(g) + O2(g) ⇌ 2SO3(g)
Calculate DG and K under the following conditions:
T = 315K, p(SO2)=0.1 atm,
DHo298 = -196.6 kJ
p(O2) = 0.2 atm, p(SO3) = 10 atm.
DGo298 = -140.0 kJ
DGo
DG315 =
315 + RT ln Q
DGo315 (-196.6) - (315)(-0.1896)
= -136.9 kJ
Q = (10)2/(0.1)2(0.2) = 50000
DSo298 = -189.6 J/K
p(SO 3 )2
Q
2
p(SO 2 ) p(O 2 )
DG315 = -136.9 + (0.008314)(315) ln (50000)
= -108.5 kJ
K315 = e
Watkins
136.9/(0.008314*315)
= 4.99×1022
Chem 1422, Chapter 19
61
2SO2(g) + O2(g) ⇌ 2SO3(g)
Summary
T
K
298
DHoT
kJ
-197
DSoT
J/K
-190
DGoT
kJ
-140
315
~ -197 ~ -190 ~ -137 ~ -109
5.0×1022
350
~ -197 ~ -190 ~ -130
-
2.7×1019
1037
~ -197 ~ -190
-
~ 1.0
~0
DGT
kJ
-
KT
(none)
3.5×1024
Le Chatelier: as the temperature of this exothermic
reaction increases, K decreases.
Watkins
Chem 1422, Chapter 19
62
C2H5OH(l)
The forward reaction is
vaporization. The
reverse reaction is
condensation.
C2H5OH(g)
C2H5OH
liquid
DHof
kJ/mol
-278
DGof
kJ/mol
-175
So
J/mol.K
161
vapor
-235
-169
283
(all values at 298K)
DHovap,298 = (-235) – (-278) = +43 kJ/mol
vaporization is endothermic
DSovap,298 = (283) – (161) = +122 J/mol.K
vaporization increases disorder
DGovap,298 = (-169) – (-175) = +6 kJ/mol
vaporization is not spontaneous at 298K
condensation is spontaneous at 298K
Watkins
Chem 1422, Chapter 19
63
C2H5OH(l)
The forward reaction is
vaporization. The
reverse reaction is
condensation.
C2H5OH(g)
DHovap,298 = +43 kJ/mol
DSovap,298 = +122 J/mol.K
DGovap,298 = +6 kJ/mol
Normal Boiling Point: the temperature T at
which a pure liquid is in equilibrium with its
vapor at 1 atm.
Standard conditions: Kp,T = pvap = 1
DGovap,T = 0 ≈ DHovap,298 – TDSovap,298
T ≈ 43/0.122 = 352K = 79 oC
experimental Tb = 78.29 oC
Watkins
Chem 1422, Chapter 19
64
C2H5OH(l)
The forward reaction is
vaporization. The
reverse reaction is
condensation.
C2H5OH(g)
DHovap,298 = +43 kJ/mol
DSovap,298 = +122 J/mol.K
DGovap,298 = +6 kJ/mol
Kp = pvap
At its normal boiling point (352K), the vapor
pressure of ethanol is 1 atm.
What is the equilibrium vapor pressure pvap of
ethanol at 298K?
Kp = pvap = e
Watkins
-DGovap,298/RT
= 0.089 atm (67 torr)
Chem 1422, Chapter 19
65
Summary
Data in Appendix C for a given substance (at 298K):
DHof,298, DGof,298, So298
Calculate for any reaction at 298K:
DHorx,298 = SmprodDHof,298 – SnreactDHof,298
DGorx,298 = SmprodDGof,298 – SnreactDGof,298
DSorx,298 = SmprodSo298 – SnreactSo298
Watkins
Chem 1422, Chapter 19
66
Summary
Calculated for any reaction at any temperature:
DGorx,298 = DHorx,298 – 298DSorx,298
DGorx,T = DHorx,T – TDSorx,T ≈ DHorx,298 – TDSorx,298
DGorx,T = 0 and KT = 1
at T = DHorx,T/DSorx,T ≈ DHorx,298/DSorx,298
DGrx,T = DGorx,T + RT ln QT
Calculated for any reaction at equilibrium: DGrx,T = 0
KT = e -DGorx,T/RT ≈ e DSorx,298/R e -DHorx,298/RT
Watkins
Chem 1422, Chapter 19
67