Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Gene expression wikipedia , lookup
Expression vector wikipedia , lookup
Magnesium transporter wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Genetic code wikipedia , lookup
Point mutation wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Interactome wikipedia , lookup
Biochemistry wikipedia , lookup
Metalloprotein wikipedia , lookup
Western blot wikipedia , lookup
Protein purification wikipedia , lookup
Protein–protein interaction wikipedia , lookup
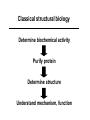
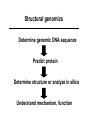
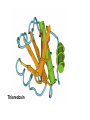
Protein Structure & Modeling Biology 224 Instructor: Tom Peavy Nov 18 & 23, 2009 <Images adapted from Bioinformatics and Functional Genomics by Jonathan Pevsner> Classical structural biology Determine biochemical activity Purify protein Determine structure Understand mechanism, function Structural genomics Determine genomic DNA sequence Predict protein Determine structure or analyze in silico Understand mechanism, function Protein function and structure Function is often assigned based on homology. However, homology based on sequence identity may be subtle. Consider RBP and OBP: these are true homologs (they are both lipocalins, sharing the GXW motif). But they are distant relatives, and do not share significant amino acid identity in a pairwise alignment. Protein structure evolves more slowly than primary amino acid sequence. RBP and OBP share highly similar three dimensional structures. Principles of protein structure Primary amino acid sequence Secondary structure: a helices, b sheets Tertiary structure: from X-ray, NMR Quaternary structure: multiple subunits Protein secondary structure Protein secondary structure is determined by the amino acid side chains. Myoglobin is an example of a protein having many a-helices. These are formed by amino acid stretches 4-40 residues in length. Thioredoxin from E. coli is an example of a protein with many b sheets, formed from b strands composed of 5-10 residues. They are arranged in parallel or antiparallel orientations. Myoglobin (John Kendrew, 1958) Thioredoxin Secondary structure prediction Chou and Fasman (1974) developed an algorithm based on the frequencies of amino acids found in a helices, b-sheets, and turns. Proline: occurs at turns, but not in a helices. GOR (Garnier, Osguthorpe, Robson): related algorithm Modern algorithms: use multiple sequence alignments and achieve higher success rate (about 70-75%) Secondary structure prediction Web servers: GOR4 Jpred NNPREDICT PHD Predator PredictProtein PSIPRED SAM-T99sec Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2] Comparative modeling (based on homology) [3] Ab initio (de novo) prediction Experimental approaches to protein structure [1] X-ray crystallography -- Used to determine 80% of structures -- Requires high protein concentration -- Requires crystals -- Able to trace amino acid side chains -- Earliest structure solved was myoglobin [2] NMR -- Magnetic field applied to proteins in solution -- Largest structures: 350 amino acids (40 kD) -- Does not require crystallization Access to PDB through NCBI Molecular Modeling DataBase (MMDB) Cn3D (“see in 3D” or three dimensions): structure visualization software Vector Alignment Search Tool (VAST): view multiple structures Additional web-based sites to visualize structures Swiss-PDB Viewer Chime RasMol MICE VRML Structural Classification of Proteins (SCOP) SCOP describes protein structures using a hierarchical classification scheme: Classes Folds Superfamilies (likely evolutionary relationship) Families Domains Individual PDB entries http://scop.mrc.lmb.cam.ac.uk/scop/ Approaches to predicting protein structures There are about >20,000 structures in PDB, and about 1 million protein sequences in SwissProt/ TrEMBL. For most proteins, structural models derive from computational biology approaches, rather than experimental methods. The most reliable method of modeling and evaluating new structures is by comparison to previously known structures. This is comparative modeling. An alternative is ab initio modeling. Approaches to predicting protein structures obtain sequence (target) fold assignment comparative modeling ab initio modeling build, assess model Comparative modeling of protein structures [1] Perform fold assignment (e.g. BLAST, CATH, SCOP); identify structurally conserved regions [2] Align the target (unknown protein) with the template. This is performed for >30% amino acid identity over a sufficient length [3] Build a model [4] Evaluate the model Errors in comparative modeling Errors may occur for many reasons [1] Errors in side-chain packing [2] Distortions within correctly aligned regions [3] Errors in regions of target that do not match template [4] errors in sequence alignment [5] use of incorrect templates Comparative modeling Many web servers offer comparative modeling services. Examples are SWISS-MODEL (ExPASy) Predict Protein server (Columbia) WHAT IF (CMBI, Netherlands) Ab initio protein structure prediction Ab initio prediction can be performed when a protein has no detectable homologs. Protein folding is modeled based on global free-energy minimum estimates. The “Rosetta Stone” methods was applied to sequence families lacking known structures. For 80 of 131 proteins, one of the top five ranked models successfully predicted the structure within 6.0 Å RMSD (Bonneau et al., 2002).