Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Gene expression wikipedia , lookup
Non-coding DNA wikipedia , lookup
Transcriptional regulation wikipedia , lookup
Genomic imprinting wikipedia , lookup
Ridge (biology) wikipedia , lookup
Homology modeling wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Community fingerprinting wikipedia , lookup
Genome evolution wikipedia , lookup
Genetic code wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Gene expression profiling wikipedia , lookup
Gene regulatory network wikipedia , lookup
Molecular evolution wikipedia , lookup
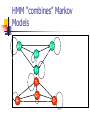
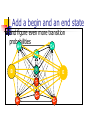
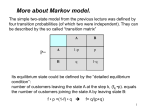
Algorithms for Finding Genes Rhys Price Jones Anne R. Haake There’s something about Genes The human genome consists of about 3 billion base pairs Some of that can be transcribed and translated to produce proteins Most of it does not What is it that makes some sequences of DNA encode protein? The Genetic Code Pick a spot in your sequence Group nucleotides from that point on in triplets. Each triplet encodes for an amino acid, or a “stop”, or a “start” TGA, TAG, TAA code for “stop” But there are exceptions! Complications In the 1960s it was thought that a gene was a linear structure of nucleotides directly linked in triplets to amino acids in the resulting protein This view did not last long with the discovery in the late 1960s of genes within genes and overlapping genes In 1977, split genes were discovered Introns and Exons Most eukaryotic genes are interrupted by non-coding sections and broken into pieces called exons. The interrupting sequences are called introns Some researchers have used a biological approach and searched for splicing sites at intron-exon junctions Catalogs of splice sites were created in the 1980s But unreliable Can you tell if it’s a gene? What is it about some sections of DNA that make them encode for proteins? Compare TTCTTCTCCAAGAGCAGGGCTTAATTCTATGCTTCCAGGCGAAAGACTGC ATGGCTAACAAAGCAACGCCTAACACATTCCTAAGCAATTGGCTTGCACC GTCTTCCCGAGGGTGTTTCTCCAATGGAAAGAGGCGTCGCTGGGCACCC GCCGGGAACGGCCGGGTGACCACCCGGTCATTGTGAACGGAAGTTTCG AGA Is there anything that distinguishes the second from the first? We’ll return to this issue Before we embark on the biological problem We will look at an easier analogy Sometimes called the Crooked Casino Basic idea: sequence of values generated by one or more processes from the values, try to deduce which process produced which parts of the sequence A useful analogy The loaded die I rolled a die 80 times and got 2363662651611344141666324264633142224 2145353556233136631321454662631116546 622542 Some of the time it was a fair die Some of the time the die produced 6s half the time and 1-5 randomly the rest of the time Confession (append (fair 20) (loaded 10) (fair 20) (loaded 10) (fair 20)) (define fair (lambda (n) (if (zero? n) '() (cons (1+ (random 6)) (fair (1- n)))))) (define loaded (lambda (n) (if (zero? n) '() (cons (if (< (random 2) 1) 6 (1+ (random 5))) (loaded (1- n)))))) 2363662651611344141666324264633142224 2145353556233136631321454662631116546 622542 There are no computer algorithms to recognize genes reliably Statistical approaches Similarity-based approaches Others Similarity Methods Searching sequences for regions that have the “look and feel” of a gene (Dan Gusfield) An early step is the investigation of ORFs (in Eukaryotes) look for possible splicing sites and try to assemble exons Combine sequence comparison and database search, seeking similar genes Statistical Approaches Look for features that appear frequently in genes but not elsewhere As for the loaded die sequence above, that is not 100% reliable But then, what is? This is Biology and it’s had billions of years to figure out how to outfox us! If you want to learn about nature, to appreciate nature, it is necessary to understand the language that she speaks in. She offers her information only in one form; we are not so unhumble as to demand that she change before we pay any attention. (R. Feynman) Relative Frequency of Stop Codons 3 of the possible 64 nucleotide triplets are stop codons We would thus expect stop codons to form approximately 5% of any stretch of random DNA, giving an average distance between stop codons of about 20 codons Very long stretches of triplets containing no stop codons (long ORFs) might, therefore, indicate gene possibilities Rather like looking for regions where my die was fair By seeking a shortage of 6s Similarly, there is (organism-specific) codon bias of various kinds, and this can be exploited by gene-finders People and programs have looked for such bias features as: regularity in codon frequencies regularity in bicodon frequencies periodicity homogeneity vs. complexity CpG Islands CG dinucleotides are often written CpG to avoid confusion with the base pair C-G In the human genome CpG is a rarer bird than would be expected in purely random sequences (there are chemical reasons for this involving methylation) In the start regions of many genes, however, the methylation process is suppressed, and CpG dinucleotides appear more frequently than elsewhere Rather like looking for stretches where my die was loaded By seeking a plethora of 6s But now we’ll look for high incidences of CpG How can we detect shortage or plethora of CpGs? Markov Models Assume that the next state depends only on the previous state If the states are x1 x2 ... xn We have a matrix of probabilities pij pij is the probability that state xj will follow xi Transition matrices CpG islands A C A 18 27 C 17 37 G 16 34 T 8 35 G 43 27 37 38 T 12 19 13 18 elsewhere A C A 30 20 C 32 30 G 25 25 T 18 24 G 28 8 30 29 T 21 30 20 29 (adapted from Durbin et al, 1998) How do we get those numbers? empirically lots of hard wet and dry lab work We can think of these numbers as parameters of our model later we’ll pay more attention to model parameters For our loaded die example we have 0 means non-6 loaded 0 6 0 50 50 unloaded 0 0 83 6 83 6 50 50 6 17 17 Discrimination Given a sequence x we can calculate a log odds score that it was generated by one of two models. S(x) = log (P(x | model A) / P(x | model B)) = sum of log((prob xi-1 to xi in A) / (prob xi-1 to xi in B)) Demonstration (define beta (lambda (x y) (cond ((and (= x ((and (= x ((and (< x ((and (< x 6) 6) 6) 6) (< (= (< (= y y y y 6)) 6)) 6)) 6)) (log (log (log (log (/ (/ (/ (/ .83 .17 .83 .17 .5))) .5))) .5))) .5)))))) (define s (lambda (l) (cond ((null? (cdr l)) 0) (else (+ (beta (car l) (cadr l)) (s (cdr l))))))) Similar Analysis for CpG Islands Visit the code (show mary) (show gene) (scorecpg mary) (scorecpg gene) Sliding Window (slidewindow 20 mary) (show (classify mary)) Hidden Markov Models The Markov model we have just seen could be used for locating CpG islands For example you could calculate the log odds score of a moving window of, say 50 nucleotides Hidden Markov Models try to combine the two models in a single model For each state, say x, in the Markov model, we imagine two states xA and xB for the two models A and B Both new states “emit” the same character But we don’t know whether it was CA or CB that “emitted” the nucleotide C HMMs Hidden Markov Models are so prevalent in bioinformatics, we’ll just refer to them as HMMs Given a sequence of “emitted signals” -- the observed sequence and a matrix of transition probabilities and emission probabilities For a path of hidden states, calculate the conditional probability of the sequence given the path Then try to find an optimal path for the sequence such that the conditional probability is maximized HMM “combines” Markov Models A+ 27 17 16 C+ 34 27 43 8 18 35 G+ 37 38 13 12 19 T+ 29 21 T- 37 18 30 20 29 30 A- 18 25 32 28 G30 8 24 25 20 30 C- Must be able to move between models Figure out these transition probabilities A+ C+ G+ T+ TGA- C- Add a begin and an end state and figure even more transition probabilities A+ C+ G+ T+ B E TGA- C- Basic Idea Each state emits a nucleotide (except B and E) A+ and A- both emit A C+ and C- both emit C You know the sequence of emissions You don’t know the exact sequences of states Was it A+ C+ C+ G- that produced ACCG Or was it A- C- C+ G- You want to find the most likely sequence Parameters of the HMM How do we know the transition probabilities between hidden states? empirical, hard lab work as above for the CpG island transition probabilities imperfect data will lead to imperfect model Is there an alternative? Machine Learning Grew out of other disciplines, including statistical model fitting Tries to automate the process as much as possible use very flexible models lots of parameters let the program figure them out Based on ... known data and properties training sets Induction and inference Bayesian Modeling Understand the hypotheses (model) Assign prior probabilities to the hypotheses Use Bayes’ theorem (and the rest of “probability calculus”) to evaluate posterior probabilities for the hypotheses in light of actual data Neural Networks Very flexible Inputs Thresholds Weights Output Variations: additional layers loops Training Neural Networks Need set of inputs and desired outputs Run the network with a given input Compare to desired output Adjust weights to improve Repeat... Training HMMs Similar idea Begin with guesses for the parameters of the HMM Run it on your training set, and see how well it predicts whatever you designed it for Use the results to adjust the parameters Repeat... Hope that When you provide brand new inputs The trained network will produce useful outputs Some gene finder programs GRAIL – neural net discriminant Genie – HMM GENSCAN – “Semi Markov Model” GeneParser – neural net FGENEH etc (Baylor) – Classification by Linear Discrimination Analysis (LDA) a well-known statistical technique GeneID – rule-based