Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
5-HT2C receptor agonist wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
Discovery and development of cyclooxygenase 2 inhibitors wikipedia , lookup
Discovery and development of antiandrogens wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Cannabinoid receptor antagonist wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Prescription costs wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Pharmacognosy wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Drug discovery wikipedia , lookup
Nicotinic agonist wikipedia , lookup
Drug design wikipedia , lookup
Plateau principle wikipedia , lookup
Drug interaction wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Psychopharmacology wikipedia , lookup
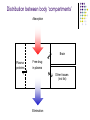
Pharmacokinetics and pharmacodynamics Sara Williamson Pharmacist Fulbourn Hospital Pharmacokinetics ‘What the body does to the drug’ Pharmacodynamics ‘What the drug does to the body’ Absorption and distribution Metabolism and excretion Plasma conc Time Total dose reaching the systemic circulation = area under the curve Bioavailability = proportion of total dose administered that reaches the systemic circulation iv Plasma conc sc oral Time Absorption - factors affecting Route of administration (e.g. IV, IM, oral) Formulation factors (e.g. MR tablets, depot injections) For oral dosage forms Food in stomach GI motility Drug interactions in GI tract Specific transporters (may be saturable) First-pass metabolism (Enterohepatic recirculation) Distribution Approximates to ‘two-compartment’ model Central compartment (plasma) Drug absorbed into this compartment Drug eliminated from this compartment Peripheral compartment (tissues including target organs) These compartments in dynamic equilibrium Distribution between body ‘compartments’ Absorption Brain Plasma proteins Free drug in plasma Other tissues (incl fat) Elimination Distribution - factors affecting Lipid solubility of drug (crossing blood brain barrier, sequestering in fat) Binding to plasma proteins (albumin, α1-glycoprotein) Blood flow to tissues Specific transport systems (e.g. levodopa across blood-brain barrier) Apparent volume of distribution Vd Obtained by measuring concentration of drug in plasma after IV injection If high (>1 l/kg) drug has high affinity for tissues outside body water (e.g. brain, fat) Chlorpromazine Vd 21 l/kg Paroxetine Vd 12 l/kg If low (c0.05 l/kg) drug has high affinity for plasma proteins Levothyroxine Highly protein-bound drugs >95% bound Diazepam Amitriptyline Warfarin Levothyroxine Furosemide 90-95% bound Phenytoin Valproate Propranolol Protein-binding - factors affecting Low plasma albumin levels Displacement from binding site by another drug (usually a transient effect) e.g. aspirin displacing warfarin Saturability of protein-binding within therapeutic range e.g. age, malnutrition, chronic liver disease e.g. valproate NB Plasma assays usually report ‘total’ rather than ‘free’ drug levels Drug metabolism Main site - liver (also some in GI wall, plasma, lung, kidney) Phase I metabolism (oxidation/ reduction/ hydrolysis/ N-demethylation) to produce more polar compounds Phase II metabolism (conjugation, usually with glucuronic acid) to produce inactive compounds Some phase I metabolites have pharmacological activity (e.g. norfluoxetine, desmethyldiazepam) Cytochrome P450 family Responsible for oxidative metabolism of most psychotropics Dominant isoenzyme CYP 3A4, also 2D6, 1A2, et al Genetic variation in enzyme activity (?pharmacogenetic testing) Drugs may affect enzyme activity, or compete as substrates Reduced activity in older people, liver disease Some enzyme-inducers Carbamazepine (↓risperidone, OCs, + autoinduction) Phenytoin (↓TCAs) Phenobarbitone (↓warfarin) Smoking (↓clozapine, olanzapine) Alcohol – chronic (↓phenytoin) St John’s wort (↓ciclosporin) Rifampicin (↓methadone) Some enzyme-inhibitors Erythromycin (↑carbamazepine) Fluvoxamine (↑clozapine) Valproate (↑lamotrigine) Ciprofloxacin (↑warfarin) Cimetidine (↑venlafaxine) Grapefruit juice (↑sertraline) Excretion Main site - kidneys (also bile, expired air, breast milk, saliva, tears) Drug may be excreted Unchanged - e.g. lithium, amisulpride As water-soluble metabolite Excretion - factors affecting Renal impairment Age Acute Chronic Assume every elderly patient has at least some renal impairment Drugs that reduce renal blood flow e.g. NSAIDs First-order kinetics: elimination half-life 110 100 Concentration (m g/L) 90 80 70 60 50 40 30 20 10 0 0 1 2 3 4 5 6 7 8 9 Tim e (hours) Half-life is the time taken for the concentration of drug in blood to fall by 50% (in this case, 1 hour) After 5 half-lives, concentration has fallen by 95% Elimination half-life +/- variation within individuals and between individuals Differences in plasma proteins, fat:lean, hepatic function, renal function May change on chronic dosing Enzyme induction/inhibition Also consider effect of active metabolites Non-first-order kinetics Rate of elimination is not proportional to plasma concentration Zero-order Elimination rate is constant (e.g. alcohol); half-life is variable Capacity-limited clearance Metabolic enzymes may be saturated at high concentrations so zero-order (slower), becoming first-order as concentrations come down (e.g. phenytoin) Consider in overdoses Withdrawal/discontinuation Generally, discontinuation effects more pronounced in drugs with faster elimination (shorter half-life) Paroxetine vs fluoxetine Lorazepam vs diazepam Heroin vs methadone Multiple doses For some drugs, it is appropriate to allow the plasma concentration to reduce substantially before the next dose is given (e.g. hypnotics) More usually, repeated doses are given to achieve a ‘steady-state’ plasma concentration. Usual dosing interval approximately 1 half-life Time to reach steady state = 4-5 half-lives Multiple doses - e.g. hypnotics Effective threshold Plasma conc Time Multiple dosing: 10mg / 12hrs At steady state amount administered = amount eliminated between doses Cavss Cp Rising phase of the curve is governed by the rate of elimination (half-life = 12 hrs) Time Cavss 20mg / 12hrs Cp 10mg / 12hrs Cavss Increasing the dose makes Cavss higher but time to steady state remains the same Time Cavss 10mg / 12hrs 20mg / 24hrs Increasing the dose and increasing the interval Cavss remains the same but fluctuation in Cp is more Therapeutic drug monitoring Useful for Drugs with low therapeutic index Drugs where effect relates to plasma concentration In psychiatric practice, mostly used for Lithium Clozapine Carbamazepine Phenytoin (Valproate?) TDM - timing issues Sample collection! Is the plasma level at steady state? At least 5 half-lives More for drugs that induce their own metabolism (e.g. carbamazepine) Is it a ‘trough’ sample? Sample immediately before next dose, or at least 12 hours after the previous dose Depot antipsychotics First-generation antipsychotics: esters of active drug, dissolved in oil Absorption slow Peak concentration after Rate-limiting half-life c14-21 days Time to reach steady state c8-12 weeks Risperdal® Consta: aqueous suspension of microparticles Extremely delayed onset of absorption - 3 weeks each time Peaks 4-5 weeks after injection Risperidone released for up to 8 weeks after injection Fluphenazine decanoate 6-48 hours Pipotiazine palmitate 9-10 days Pharmacodynamics ‘What the drug does to the body’ It includes both the desired action of the drug and undesired actions such as adverse effects and interactions Mechanisms of drug action in the body Binding to receptors Interaction with enzymes Interaction with ion channels Interaction with carrier proteins Interaction with structural proteins Cellular membrane disruption Chemical reaction Drug action in the body Most psychoactive drugs affect neurotransmitters: Synthesis Storage Release Reuptake Degradation Direct action at receptors Second-messenger function Binding to receptors Drugs may be Agonists Antagonists Partial agonists Efficacy is related to the size of effect Potency is related to the amount required to produce that effect Binding may be Competitive or noncompetitive Reversible or irreversible Efficacy and potency 100% Maximum effect 50% Agonist concentration (log scale) Competitive and noncompetitive binding 100% Maximum effect with competitive antagonist 50% with noncompetitive antagonist Agonist concentration (log scale) Partial agonists 100% 50% Maximum effect Full agonist Full agonist activity, adding partial agonist Partial agonist Full agonist activity adding antagonist Drug concentration (log scale) Binding to receptors Drugs may mimic the shape, charge etc of the natural neurotransmitter May compete with a natural neurotransmitter for binding at receptor Example: first-generation antipsychotics FGAs are dopamine D2 antagonists – block dopamine transmission in mesolimbic (↓ positive symptoms) Side-effects May be due to the primary action e.g. FGAs also block D2 receptors in other pathways nigrostriatal (EPSEs) tuberoinfundibular (↑prolactin) May be due to action at other receptors e.g. muscarinic antagonist (dry mouth, constipation, blurred vision, cognitive blunting) H-1 histaminic antagonist (weight gain, drowsiness) α-1 adrenergic antagonist (dizziness, ↓BP) Receptor profiles Balance of receptor affinities may improve clinical effects or reduce side-effects Example: second-generation antipsychotics SGAs have 5HT2A antagonist activity as well as D2 antagonism 5HT2A antagonism in the nigrostriatal pathway stimulates dopamine release (↓EPSEs) 5HT2A antagonism in the mesocortical pathway stimulates dopamine release (may improve affective, cognitive and negative symptoms) Transport carriers Move neurotransmitter from the synapse back into the presynaptic terminal Need an energy source eg Na+/K+ ATPase May also involve an ion eg Na+ which increases the affinity of the transporter for the neurotransmitter Example: SSRIs Reuptake pump removes serotonin from the synapse – blocked by SSRIs so ↑serotonin But initial effects at pre-synaptic 5HT1A receptors reduces postsynaptic serotonin release Down-regulation of pre-synaptic receptors allows increase in serotonin (delayed response to antidepressant) Side-effects include nausea (5HT2A) and sexual dysfunction (5HT3) Enzymes Involved in the synthesis and degradation of neurotransmitters May be activated or inhibited by drugs Example: monoamine oxidase inhibitors Monoamine oxidases break down serotonin/noradrenaline (MAO-A) and dopamine (MAO-B) MAOI antidepressants prevent this breakdown ‘Cheese’ reaction due to inhibition of MAO-A in gut MAOIs (e.g. phenelzine) inhibit irreversibly; moclobemide is a reversible inhibitor (RIMA) Selegiline inhibits MAO-B - ↑dopamine (in Parkinson’s) Ion channels Involved in fast-onset signals; may be voltage gated or ligand gated Example: Benzodiazepines Gamma-aminobutyric acid inhibits most neurons Benzodiazepines bind to GABA-A receptors, enhancing the effect of GABA Example: Memantine Glutamate stimulates most neurons; in Alzheimer’s, excess glutamate causes ‘background noise’ and cell damage Memantine blocks NDMA receptors (one type of glutamate receptor) GABA and glutamate Some more examples: Valproate may increase GABA function and reduce glutamate function Carbamazepine may increase GABA function Lamotrigine may inhibit the release of glutamate G protein-linked receptors and second messengers Neurotransmitter binds to receptor Receptor is changed so G protein can bind G protein binds and is changed so an enzyme can bind Enzyme binds and synthesizes a second messenger (e.g. c-AMP) This second messenger may act on ion channels, other enzymes or gene transcription within the cell Lithium Mechanism of action? Lithium may prevent the G protein from binding to the activated receptor; or May prevent the enzyme from binding to the G protein and therefore preventing a second messenger being produced; or May interfere with gene expression as modulated by protein kinase C, regulating growth factors and neuronal plasticity