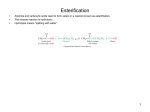
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Evolution of metal ions in biological systems wikipedia , lookup
Peptide synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Metalloprotein wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Mitochondrion wikipedia , lookup
Proteolysis wikipedia , lookup
Genetic code wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Specialized pro-resolving mediators wikipedia , lookup
Butyric acid wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Biochemistry wikipedia , lookup
Citric acid cycle wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
BC 23 Fuel Oxidation and Generation of ATP Liver transaminases measured in blood are AST (SGOT) and ALT (SGPT); elevation in serum reflects damage of liver cell PMs; transaminases catalyze transfer of nitrogen group of amino acid to acceptor α-keto acid o For AST, aspartate donates nitrogen to α-ketoglutarate, forming oxaloacetate and glutamate o For ALT, alanine donates nitrogen to α-ketoglutarate, forming pyruvate and glutamate o Measured by loss of NADH in reaction mix During moderate-intensity exercise, decreases in insulin and increases in insulin counterregulatory hormones (epinephrine and norepinephrine) increase adipose tissue lipolysis; muscles provided with supply of fatty acids in blood that can be used as fuel During fasting, muscle tissue, liver, and other tissues oxidize fatty acids as fuel; after overnight fasting, 60-70% of energy supply derived from oxidation of fatty acids Fatty Acids as Fuels Fatty acids oxidized as fuels principally long-chain fatty acids released from adipose tissue triacylglycerol stores between meals, during overnight fasting, and during periods of increased fuel demand (exercising) o Adipose tissue triacylglycerols derived from dietary lipids or synthesized in liver o Long-chain fatty acids (palmitate, oleate, and stearate) highest in dietary lipids and synthesized, so major fuel oxidation Between meals, decreased insulin and increased insulin counterregulatory hormones activate lipolysis Free fatty acids transported bound to serum albumin In tissues, energy derived from oxidation of fatty acids to acetyl-CoA in β-oxidation o Most of enzymes involved in fatty acid oxidation present as 2-3 isoenzymes, which have different but overlapping specificities for chain length of fatty acid o Acetyl-CoA produced from fatty acid oxidation principally oxidized in TCA cycle or converted to ketone bodies in liver Huge majority of fat in diet is triacylglycerols Most common dietary fatty acids are saturated long-chain fatty acids palmitate (C16) and stearate (C18), monounsaturated fatty acid oleate (C18:1), and polyunsaturated essential fatty acid linoleate (C18:2) o Animal fat contains principally saturated and monounsaturated long-chain fatty acids o Vegetable oils contain linoleate and some longer chain and polyunsaturated fatty acids; small amounts of branched-chain and odd-chain-length fatty acids o Medium-chain-length fatty acids principally in dairy fat and vegetable oils Adipose tissue triacylglycerols contain fatty acids synthesized in liver, principally from excess calories (glucose) Long-chain fatty acids toxic to cells because they can disrupt hydrophobic bonding between amino acid side chains in proteins; transported in blood and in cells bound to protein During times of metabolic need, long-chain fatty acids released from adipose tissue triacylglycerols by lipases o Enter cells by saturable transport process and by diffusion through PM o Fatty acid-binding protein binds fatty acid intracellularly; facilitates transport to mitochondrion o Free fatty acid concentration in cells extremely low Fatty acids must be activated to acyl-CoA derivatives before they can participate in β-oxidation and other metabolic pathways o Activation involves thiokinase; uses ATP (forms AMP and PPi); subsequent cleavage of PPi drives reaction o Acyl-CoA synthase that activates long-chain fatty acids present in ER, outer mitochondrial membranes, and peroxisomal membranes; no activity towards longer or shorter chains o Synthetase for activation of very long-chain fatty acids present in peroxisomes o Medium-chain-length fatty acid-activating enzyme only in mitochondrial matrix of liver and kidneys Carnitine serves as carrier for activated long-chain fatty acyl group across inner mitochondrial membrane o Carnitine acyl transferases able to reversibly transfer activated fatty acyl group from CoA to carnitine to form acylcarnitine ester o CPTI transfers long-chain fatty acyl groups from CoA to carnitine on outer mitochondrial membrane o Fatty acylcarnitine crosses inner mitochondrial membrane with aid of translocase o Fatty acyl group transferred back to CoA by CPTII; carnitine released returns to cytosolic side of mitochondrial membrane by same translocase that brings it over o Long-chain fatty acyl-CoA substrate for β-oxidation in mitochondrial matrix Carnitine can be obtained from diet or synthesized from side chain of lysine (begins in skeletal muscle, completed in liver); requires vitamin C Most carnitine in body stored in skeletal muscle Classical CPTII deficiency – characterized by adolescent-to-adult onset of recurrent episodes of acute myoglobinuria precipitated by prolonged exercise or fasting; patient weak and may be somewhat hypoglycemic with diminished ketosis (hypoketosis); metabolic decompensation not severe o Lipid deposits found in skeletal muscles o CPK and long-chain acylcarnitines elevated in blood o CPTII levels in fibroblasts 25% of normal; remaining CPTII activity accounts for mild effect on liver metabolism o When CPTII deficiency presents in infants, levels are <10% of normal; hypoglycemia and hypoketosis severe, hepatomegaly occurs from triacylglycerol deposits; cardiomyopathy present Carnitine deficiency found only in infants fed soy-based formula not supplemented with carnitine Riboflavin is vitamin precursor of FAD (required for acyl-CoA dehydrogenases and electron-transfer flavoproteins (ETFs) o CoQ synthesized in body; recipient in electron-transport chain for electrons passed from complexes I and II and ETFs o Supplementation of pantothenate (precursor of CoA) improves performance Oxidation of fatty acids in β-oxidation conserves energy as FAD(2H) and NADH Acetyl-CoA oxidized in TCA cycle or converted to ketone bodies β-oxidation requires (in order) acyl-CoA dehydrogenase (transfers electrons to FAD), enoyl hydratase, hydroxyacyl-CoA dehydrogenase (electrons transferred to NAD+), β-ketothiolases o Fatty acyl-CoA continues the loop until all carbons converted to acetyl-CoA (16 carbon fatty acid would produce 7 FAD(2H)s, 7 NADHs, and 8 acetyl-CoAs) o NADH and FAD(2H) passed to electron transport chain; generates 1.5 ATP for each FAD(2H) and 2.5 for each NADH (total energy yield of previous 16-C fatty acid is 28 ATP) o As fatty acyl chains shortened by consecutive cleavage of 2 acetyl units, they are transferred from enzymes that act on longer chains to those that act on shorter chains Unsaturated fats usually in cis bonds; during β-oxidation of saturated fatty acids, trans bond formed; for unsaturated fatty acids to undergo β-oxidation, the cis bond must be isomerized to trans Linoleate – essential fatty acid; only portion not needed for other processes oxidized MCAD deficiency – long-chain fatty acids metabolized by β-oxidation to medium-chain acyl-CoA, but further oxidation blocked, so medium-chain acyl group transferred back to carnitine o Acylcarnitines water-soluble and appear in blood and urine LCAD deficiency – fatty acylcarnitines accumulate in blood, but not urine Odd-chain-length fatty acids undergo β-oxidation until last spiral (5 carbons remain); cleavage by thiolase produces acetyl-CoA and propionyl-CoA; carboxylation of propionyl-CoA yields methylmalonyl-CoA, which is converted to succinyl-CoA in vitamin B12-dependent reaction (anaplerotic for TCA cycle) o Propionyl-CoA also arises from oxidation of BCAAs Acetyl-CoA formed from β-oxidation of even-chain fatty acids in liver either enters TCA cycle or converted to ketone bodies Dietary medium-chain fatty acids more water-soluble than long-chain fatty acids and not stored in adipose triacylglycerol; enter blood and pass through portal vein to liver, where they enter mitochondrial matrix by monocarboxylate transporter and activated by acyl-CoA derivatives in mitochondrial matrix Medium-chain acyl-CoAs can arise from peroxisomal oxidation pathway Medium-chain acyl-CoA synthetase has broad specificity; can bind drugs (aspirin, valproate, benzoate); once drug-CoA derivative formed, carboxyl group conjugated with glycine to form urinary excretion product Unripe fruit of ackee tree produces toxin (hypoglycin) which causes Jamaican vomiting sickness; patients develop severe hypoglycemia (often fatal); hypoglycin inhibits acyl-CoA dehydrogenase involved in β-oxidation of short-chain and medium-chain fatty acids o Because more glucose must be oxidized to compensate for decreased ability of fatty acids to be used as fuel, blood glucose levels fall to extremely low levels o Fatty acid levels rise because of decreased β-oxidation; because of this, ω-oxidation increases, and dicarboxylic acids excreted in urine o Diminished capacity to oxidize fatty acids in liver mitochondria results in decreased levels of acetyl-CoA (substrate for ketone body synthesis) Fatty acids used as fuels when released from adipose tissue triacylglycerols in response to hormones that signal fasting or increased demand o Muscle and kidney oxidize fatty acids completely to CO2 and H2O; acetyl-CoA produced by β-oxidation enters TCA cycle and passes NADH and FAD(2H) to electron transport chain o Process regulated by cells’ requirements for energy Fatty acid oxidation can be restricted by mitochondrial CoASH pool size; acetyl CoASH must enter TCA cycle or another metabolic pathway to regenerate CoASH required for formation of fatty acyl-CoA derivative from fatty acyl carnitine CPTI inhibited by malonyl-CoA synthesized in cytosol by acetyl-CoA carboxylase, which is regulated by tissuedependent mechanisms o Skeletal muscle and liver – inhibited when phosphorylated by AMP-PK During exercise (AMP level increase), AMP-PK activated and phosphorylates acetyl-CoA carboxylase, which becomes inactive; malonyl-CoA levels decrease, and CPTI activated, and βoxidation of fatty acids restores ATP homeostasis and decreases AMP levels o In liver, acetyl-CoA carboxylase activated by insulin-dependent mechanisms leading to elevated cytoplasmic citrate (allosteric activator) which promotes conversion of malonyl-CoA to palmitate in fatty acid synthesis pathway Malonyl-CoA inhibition of CPTI prevents newly synthesized fatty acids from being oxidized β-oxidation strictly aerobic pathway (requires mitochondria) Fatty acids not significant fuel for brain and not used by adipocytes Tissues that don’t use fatty acids much as fuel able to use ketone bodies instead Alternative Routes of Fatty Acid Oxidation Fatty acids not oxidized by β-oxidation enter peroxisomal β-oxidation, α-oxidation, and microsomal ω-oxidation; pathways convert unusual fatty acids to compounds that can be used as fuels or biosynthetic precursors and convert remainder to compounds that can be excreted in bile or urine During prolonged fasting, fatty acids released from adipose triacylglycerols may enter ω-oxidation or peroxisomal β-oxidation pathway, even though they have normal composition Pathways also act on xenobiotic carboxylic acids that are large hydrophobic molecules resembling fatty acids Very long-chain fatty acids oxidized exclusively by peroxisomal β-oxidation and α-oxidation pathways (chainshortening pathways); o β-oxidation 1 NADH and 1 acetyl-CoA generated for each turn of spiral; spiral turns until medium-chain acyl-CoA produced o Acetyl groups in peroxisome can be transferred from CoA to carnitine or can enter cytosol Acylcarnitines diffuse from peroxisome to mitochondria, pass through outer mitochondrial membrane, and are transported through inner mitochondrial membrane via carnitine translocase system; converted back to acyl-CoAs by carnitine:acyltransferases and enter normal pathways for β-oxidation and acetyl-CoA metabolism o Electrons pass from peroxisome to cytosol through use of shuttle system similar to mitochondria Zellweger syndrome – defective peroxisomal biogenesis; leads to developmental and metabolic phenotypes that affect principally liver and brain o Elevation of C26:0 and C26:1 fatty acid levels in plasma Refsum disease caused by deficiency in phytanoyl-CoA hydroxylase that carries out peroxisomal α-oxidation of phytanic acid; symptoms include retinitis pigmentosa, cerebellar ataxia, and chronic polyneuropathy o Phytanic acid obtained solely from diet, so placing patient on low-phytanic acid diet helps Phytanic acid and pristanic acid (long-chain branched-chain fatty acids that are degradation products of chlorophyll) oxidized in peroxisomes to branched C8 fatty acid, which is transferred to mitochondria o Phytanic acid oxidized via α-oxidation pathway; fatty acid shortened so methyl groups appear on αcarbon and can no longer interfere with oxidation of β-carbon; peroxisomal β-oxidation then goes When medium-chain length reached, fatty acid transferred to mitochondrion as carnitine derivative and β-oxidation resumed ω-oxidation in ER; first step uses P450, O2, and NADPH o Dicarboxylic acids produced by ω-oxidation can undergo β-oxidation; can enter blood and be oxidized as medium-chain fatty acids or be excreted in urine as medium-chain dicarboxylic acids Peroxisomal β-oxidation and α-oxidation and microsomal ω-oxidation not feedback regulated; function to decrease levels of water-insoluble fatty acids or xenobiotic compounds with fatty acid-like structure that would become toxic at high concentrations, so regulated by availability of substrate Normally ω-oxidation minor process, but in conditions that interfere with β-oxidation (such as carnitine deficiency or deficiency in enzyme of β-oxidation), ω-oxidation produces dicarboxylic acids in increased amounts, which are then excreted in urine Metabolism of Ketone Bodies Fatty acids released from adipose triacylglycerols serve as major fuel for body during fasting In liver, much of acetyl-CoA generated from β-oxidation of fatty acids used for synthesis of ketone bodies acetoacetate and β-hydroxybutyrate, which enter blood o In skeletal muscles and other tissues, ketone bodies converted back to acetyl-CoA, which is oxidized in TCA cycle with generation of ATP o Alternate fate of acetoacetate is formation of cytosolic acetyl-CoA In liver, ketone bodies synthesized in mitochondrial matrix from acetyl-CoA generated from fatty acid oxidation o Last reaction of fatty acid oxidation is reversible, so acetyl-CoA turns back to acetoacetyl-CoA for ketone body synthesis o Acetoacetyl-CoA reacts with acetyl-CoA to produce HMG-CoA, which forms acetyl-CoA and acetoacetate Acetoacetate can enter blood directly or can be reduced to β-hydroxybutyrate, which enters blood o Reaction readily reversible and interconverts ketone bodies, which exist in equilibrium ratio determined by NADH/NAD+ ratio of mitochondrial matrix (usually 1:1 ratio of ketone bodies) o Acetoacetate can spontaneously decarboxylate into CO2 and acetone; because acetone volatile, it is expired by lungs and small amount of acetone further metabolized by body Ketone bodies can be oxidized as fuels in most cells; transported to mitochondrial matrix, where βhydroxybutyrate turned back into acetoacetate, producing NADH o Acetoacetate then turned into acetyl-CoA o In mitochondria, acetoacetate activated by succinyl-CoA from TCA cycle o Liver doesn’t use ketone bodies it makes because it doesn’t have thiotransferase o Acetoacetyl-CoA cleaved to 2 molecules of acetyl-CoA by acetoacetyl-CoA thiolase (same enzyme involved in β-oxidation; acetyl-CoA sent through TCA cycle o Energy yield of 19 ATP for acetoacetate o Oxidation of β-hydroxybutyrate provides one additional NADH, so net energy yield of 1 βhydroxybutyrate is 21.5 ATP Ketogenic diets high-fat diets; used to reduce frequency of epileptic seizures in children o Also used to treat children with pyruvate dehydrogenase deficiency o Ketone bodies used as fuel by brain in absence of pyruvate dehydrogenase o Ketone bodies can also provide source of cytosolic acetyl-CoA for ACh synthesis Medium-chain triglycerides induce ketosis more effectively than long-chain triglycerides Ketone bodies can be generated from catabolism of certain amino acids (ketogenic amino acids: leucine, isoleucine, lysine, tryptophan, phenylalanine, and tyrosine); carbon skeletons catabolized to acetyl-CoA or acetoacetyl-CoA which enter pathway of ketone body synthesis in liver o Leucine and isoleucine form acetyl-CoA and acetoacetyl-CoA in other tissues too o Acetoacetyl-CoA can be used directly in cholesterol synthesis or cleaved to 2 molecules of acetyl-CoA o Cytosolic acetyl-CoA required for processes such as ACh synthesis in neuronal cells Role of Fatty Acids and Ketone Bodies in Fuel Homeostasis Fatty acids used as fuels whenever lots of them in blood (during fasting, high-fat low-carb diet, or exercise) o Decrease in insulin and increased levels of glucagon, epinephrine, or other hormones stimulate adipose tissue lipolysis and fatty acids increase in blood 3-4 hours after meal and progressively increase with time of fasting up to 2-3 days o In liver, rate of ketone body synthesis increases as supply of fatty acids increase; blood level of ketone bodies increases because utilization by skeletal muscles decreases o After 2-3 days starvation, ketone bodies rise to level in blood that enables them to enter brain cells, where they are oxidized, reducing amount of glucose required by brain (become main fuel source for brain energy requirements) o Reduction in glucose requirements spares skeletal muscle protein, which is major source of amino acid precursors for hepatic glucose synthesis from gluconeogenesis Increased fatty acids in blood makes them preferential to glucose for use by skeletal muscles; results in relatively high NADH/NAD+ ratios, acetyl-CoA concentrations, and ATP/AMP levels o In skeletal muscles, AMP-PK adjusts concentration of malonyl-CoA so CPTI and β-oxidation operate at rate able to sustain ATP homeostasis o Glycolysis and TCA cycle decreased by changes in concentration of allosteric regulators (decreased ADP and increased NADH, ATP, citrate, and acetyl-CoA) Leads to glucose 6-P accumulation, which inhibits hexokinase, decreasing rate of entry of glucose into glycolysis o Above facilitated by decrease in insulin concentration Children more prone to ketosis than adults because bodies enter fasting state more rapidly; mild pediatric infections that cause anorexia and vomiting most common causes of ketosis in kids o Mild ketosis observed in children after prolonged exercise Skeletal muscles, heart, liver use fatty acids during fasting and under other conditions that increase fatty acids in blood; brain and intestinal mucosa use ketone bodies to greater extent o Intestinal mucosa won’t use fat because it transports it, not uses it o Adipocytes store fat, so won’t use fatty acids, but will use ketone bodies o Ketone bodies cross placenta and can be used by fetus o Liver and RBCs only things that don’t use ketone bodies Decreased insulin/glucagon ratio results in decreased malonyl-CoA levels; activates CPTI, allowing use of fatty acyl-CoA in β-oxidation o When oxidation of fatty acyl-CoA generates enough NADH and FAD(2H) to supply ATP needs of liver, acetyl-CoA diverted from TCA cycle to ketogenesis and oxaloacetate in TCA cycle diverted into gluconeogenesis; regulated by NADH/NAD+ ratio (high during β-oxidation) Prolonged fasting means increased transcription of gene fo mitochondrial HMG-CoA synthase, facilitating high rates of ketone body production LCAD deficiency rare disorder; LCAD activity contained in mitochondrial trifunctional protein that catalyzes 3 steps in oxidation of long-chain fatty acids o Generalized mitochondrial trifunctional protein deficiency can cause wide clinical spectrum of disease Acetylation and Regulation of Fatty Acid Oxidation Histone acetyltransferases (HATs) catalyze histone acetylation leading to histone dissociation from DNA Freed DNA binds factors important for transcription to occur Acetylation of LCAD reduces its activity Sirtuins (SIRT) – NAD+-dependent protein deacetylases; catalyze reaction that splits NAD+ and deacetylates protein target SIRT3 localized to mitochondrial matrix; key regulator of fatty acid oxidation in mitochondria o SIRT3 expression upregulates during fasting in liver; primary target of deacetylation is LCAD, activating it