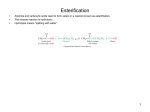
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Evolution of metal ions in biological systems wikipedia , lookup
Peptide synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Metalloprotein wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Mitochondrion wikipedia , lookup
Proteolysis wikipedia , lookup
Genetic code wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Specialized pro-resolving mediators wikipedia , lookup
Butyric acid wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Biochemistry wikipedia , lookup
Citric acid cycle wikipedia , lookup
Glyceroneogenesis wikipedia , lookup