Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Lipid signaling wikipedia , lookup
Mitochondrion wikipedia , lookup
Peptide synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Metalloprotein wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Genetic code wikipedia , lookup
Specialized pro-resolving mediators wikipedia , lookup
Proteolysis wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Butyric acid wikipedia , lookup
Citric acid cycle wikipedia , lookup
Biosynthesis wikipedia , lookup
Biochemistry wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
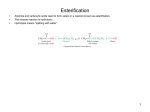
LIPID MOBILIZATION In the postabsorptive state, fatty acids can be released from adipose tissue to be used for energy production. Although human adipose tissue does not respond directly to glucagon, the fall in insulin activates a hormone-sensitive triacylglycerol lipase (HSL) that hydrolyzes triglycerides yielding fatty acids and glycerol. Epinephrine and cortisol also activate HSL. Glycerol may be picked up by liver and converted to dihydroxyacetone phosphate (DHAP) for gluconeogenesis,and the fatty acids are distributed to tissues that can use them. Free fatty acids are transported through the blood in association with albumin. Lipolysis of Triglyceride in Response to Hypoglycemia and Stress FATTY ACID OXIDATION Fatty acids are oxidized in several tissues, including liver, muscle, and adipose tissue, by the pathway of β-oxidation.Neither erythrocytes nor brain can use fatty acids, and so continue to rely on glucose during normal periods of fasting. Erythrocytes lack mitochondria, and fatty acids do not cross the blood-brain barrier efficiently. After uptake by the cell, fatty acids are activated by conversion into their CoA derivatives—acyl CoA is formed. For channeling into the mitochondria, the acyl residues are first transferred to carnitine and then transported across the inner membrane as acyl carnitine. The degradation of the fatty acids occurs in the mitochondrial matrix through an oxidative cycle in which C2 units are successively cleaved off as acetyl CoA (activated acetic acid). Before the release of the acetyl groups, each CH2 group at C-3 of the acyl residue (the β-C atom) is oxidized to the keto group— hence the term β-oxidation for this metabolic pathway. Both spatially and functionally, it is closely linked to the tricarboxylic acid cycle and to the respiratory chain. [1] The first step is dehydrogenation of acyl CoA at C-2 and C-3. This yields an unsaturated 2-enoyl-CoA derivative with a trans-configured double bond. The two hydrogen atoms are initially transferred from FAD-containing acyl CoA dehydrogenase to the electron-transferring flavoprotein (ETF). ETF dehydrogenase passes them on from ETF to ubiquinone (coenzyme Q), a component of the respiratory chain [2] The next step in fatty acid degradation is the addition of a water molecule to the double bond of the enoyl CoA (hydration), with formation of β-hydroxyacyl CoA. [3] In the next reaction, the OH group at C- 3 is oxidized to a carbonyl group (dehydrogenation). This gives rise to β-ketoacyl CoA, and the reduction equivalents are transferred to NAD+, which also passes them on to the respiratory chain [4] β-Ketoacyl-CoA is now broken down by an acyl transferase into acetyl CoA and an acyl CoA shortened by 2 C atoms (“thioclastic cleavage”). Several cycles are required for complete degradation of long-chain fatty acids—eight cycles in the case of stearyl-CoA (C18:0), for example. The acetyl CoA formed can then undergo further metabolism in the tricarboxylic acid cycle , or can be used for biosynthesis. When there is an excess of acetyl CoA, the liver can also form ketone bodies. When oxidative degradation is complete, one molecule of palmitic acid supplies around 130 molecules of ATP, corresponding to an energy of 3300 kJ mol–1. This high energy yield makes fats an ideal form of storage for metabolic energy. Hibernating animals such as polar bears can meet their own energy requirements for up to 6 months solely by fat degradation, while at the same time producing the vital water they need via the respiratory chain (“respiratory water”). Fatty acid transport : The inner mitochondrial membrane has a group-specific transport system for fatty acids. In the cytoplasm, the acyl groups of activated fatty acids are transferred to carnitine by carnitine acyltransferase [1]. They are then channeled into the matrix by an acyl carnitine/ carnitine exchange. In the matrix, the mitochondrial enzyme carnitine acyltransferase catalyzes the return transfer of the acyl residue to CoA. The carnitine shuttle is the rate-determining step in mitochondrial fatty acid degradation. Malonyl CoA, a precursor of fatty acid biosynthesis, inhibits carnitine acyltransferase ,and therefore also inhibits uptake of fatty acids into the mitochondrial matrix. The most important regulator of βoxidation is the NAD+/NADH+H+ ratio. If the respiratory chain is not using any NADH+H+, then not only the tricarboxylic acid cycle but also β-oxidation come to a standstill due to the lack of NAD+. Cytoplasm mitochondria Genetic Deficiencies of Fatty Acid Oxidation Two of the most common genetic deficiencies affecting fatty acid oxidation are: .□ Medium chain acyl CoA dehydrogenase (MCAD) deficiency,primary etiology hepatic .□ Myopathic carnitine acyltransferase (CAT/CPT) deficiency, primary etiology myopathic Medium Chain Acyl CoA Dehydrogenase (MCAD) Deficiency: Non-ketotic hypoglycemia should be strongly associated with a block in hepatic β-oxidation. During fasting, hypoglycemia can become profound due to lack of ATP to support glyconeogenesis. Decreased acetyl-CoA lowers pyruvate carboxylase activity and also limits ketogenesis. Hallmarks of MCAD deficiency include: . Profound fasting hypoglycemia . Low to absent ketones . Lethargy, coma, death if untreated . Dicarboxylic acidemia and Dicarboxylic aciduria . Episode may be provoked by overnight fast in an infant . In older child, often provoked by illness (flu) that causes loss of appetite and vomiting . Primary treatment: IV glucose . Prevention: frequent feeding, high-carbohydrate, low-fat diet Carnitine Acyltransferase (CAT/CPT) Deficiency (Myopathic Form): Although all tissues with mitochondria contain carnitine acyltransferase, the most common form of this genetic deficiency is myopathic and due to a defect in the musclespecific CAT/CPT gene. Hallmarks of this disease include: . Muscle aches; mild to severe weakness . Rhabdomyolysis, myoglobinuria, red urine . Episode provoked by prolonged exercise especially after fasting, cold, or associated stress . Symptoms may be exacerbated by high-fat, low-carbohydrate diet . Muscle biopsy shows elevated muscle triglyceride detected as lipid droplets in cytoplasm . Primary treatment: cease muscle activity; give glucose Minor pathways of fatty acid degradation Most fatty acids are saturated and even-numbered. They are broken down via βoxidation .In addition, there are special pathways involving degradation of unsaturated fatty acids ,degradation of fatty acids with an odd number of C atoms . A. Degradation of unsaturated fatty acids Unsaturated fatty acids usually contain a cis double bond at position 9 or 12 e. g., linoleic acid (18:2; 9,12). As with saturated fatty acids, degradation in this case occurs via β-oxidation until the C-9-cis double bond is reached. Since enoyl-CoA hydratase only accepts substrates with trans double bonds, the corresponding enoyl-CoA is converted by an isomerase from the cis-∆3, cis- ∆6 isomer into thetrans-∆3,cis-∆6 isomer [1]. Degradation by β-oxidation can now continue until a shortened trans-∆2, cis-∆4 derivative occurs in the next cycle. This cannot be isomerized in the same way as before, and instead is reduced in an NADPH-dependent way to the trans-∆3 compound[2]. After rearrangement by enoyl-CoA isomerase [1], degradation can finally be completed via normal β-oxidation. B. Degradation of odd numbered fatty acids Fatty acids with an odd number of C atoms are treated in the same way as “normal” fatty acids—i. e., they are taken up by the cell with ATP dependent activation to acyl CoA and are transported into the mitochondria with the help of the carnitine shuttle and broken down there by β−oxidation . In the last step, propionyl CoA arises instead of acetyl CoA. This is first carboxylated by propionylCoA carboxylase into methylmalonylCoA, which is isomerized by mutase into succinyl CoA . Various coenzymes are involved in these reactions. The carboxylase requires biotin, and the mutase is dependent on coenzyme B12. Odd-numbered fatty acids from propionyl-CoA can therefore be used to synthesize glucose. This pathway is also important for ruminant animals, which are dependent on symbiotic microorganisms to break down their food. The microorganisms produce large amounts of propionic acid as a degradation product, which the host can channel into the metabolism in the way described. Degradation of Phosphoglycerols Although phospholipids are actively degraded, each portion of the molecule turns over at a different rate . Phospholipase A2 catalyzes the hydrolysis of glycerophospholipids to form a free fatty acid and lysophospholipid, which in turn may be reacylated by acyl-CoA in the presence of an acyltransferase. Alternatively, lysophospholipid (eg, lysolecithin ) is attacked by lysophospholipase, forming the corresponding glyceryl phosphoryl base, which in turn may be split by a hydrolase liberating glycerol 3-phosphate plus base. Phospholipases A1, A2, C, and D attack lipid bonds . Phospholipase A2 is found in pancreatic fluid and snake venom as well as in many types of cells; phospholipase C is one of the major toxins secreted by bacteria; and phospholipase D is known to be involved in mammalian signal transduction. Long-chain saturated fatty acids are found predominantly in the 1 position of phospholipids, whereas the polyunsaturated acids (eg, the precursors of prostaglandins) are incorporated more into the 2 position. Sphingolipids are Degraded in Lysosomes Most cells continually degrade and replace their membrane lipids. For each hydrolyzable bond in a phospholipid, there is a specific hydrolytic enzyme in the lysosome . Gangliosides are degraded by a set of lysosomal enzymes that catalyze the stepwise removal of sugar units, finally yielding a ceramide. A genetic defect in any of these hydrolytic enzymes leads to the accumulation of gangliosides in the cell, with severe medical consequences . CHOLESTEROL IS DERIVED ABOUT EQUALLY FROM THE DIET& FROM BIOSYNTHESIS A little more than half the cholesterol of the body arises by synthesis (about 700 mg/d), and the remainder is provided by the average diet. The liver and intestine account for approximately 10% each of total synthesis in humans. Virtually all tissues containing nucleated cells are capable of cholesterol synthesis, which occurs in the endoplasmic reticulum and the cytosol. CHOLESTEROL IS EXCRETED FROM THE BODY IN THE BILE AS CHOLESTEROL OR BILE ACIDS About 1 g of cholesterol is eliminated from the body per day. Approximately half is excreted in the feces after conversion to bile acids. The remainder is excreted as cholesterol. Coprostanol is the principal sterol in the feces; it is formed from cholesterol by the bacteria in the lower intestine. Bile Acids Are Formed From Cholesterol The primary bile acids are synthesized in the liver from cholesterol. These are cholic acid (found in the largest amount) and chenodeoxycholic acid . Primary bile acids are those synthesized by the liver. Secondary bile acids result from bacterial actions in the colon. A portion of the primary bile acids in the intestine is subjected to further changes by the activity of the intestinal bacteria. MANY HORMONES ARE MADE FROM CHOLESTEROL Adrenal Steroidogenesis All mammalian steroid hormones are formed from cholesterol via pregnenolone through a series of reactions that occur in either the mitochondria or endoplasmic reticulum of the adrenal cell. Hydroxylases that require molecular oxygen and NADPH are essential, and dehydrogenases, an isomerase, and a lyase reaction are also necessary for certain steps. There is cellular specificity in adrenal steroidogenesis. For instance, 18- hydroxylase and 19-hydroxysteroid dehydrogenase, which are required for aldosterone synthesis, are found only in the zona glomerulosa cells (the outer region of the adrenal cortex), so that the biosynthesis of this mineralocorticoid is confined to this region. Testicular Steroidogenesis Ovarian Steroidogenesis 1,25(OH)2-D3 (Calcitriol) Is Synthesized From a Cholesterol Derivative Lipid metabolic scheme 1- Electrolytes Sodium Potassium Chloride Bicarbonate 2- Renal (Kidney) Function Tests Creatinine Blood urea nitrogen 3- Liver Function Tests Total protein (serum) Albumin Globulins A/G ratio (albumin-globulin) Protein electrophoresis Urine protein Bilirubin; direct; indirect; total Aspartate transaminase (AST) Alanine transaminase (ALT) Gamma-glutamyl transpeptidase (GGT) Alkaline phosphatase (ALP) 4- Cardiac Markers H-FABP (Heart-type Fatty Acid Binding Protein) Troponin Myoglobin CK-MB B-type natriuretic peptide (BNP) 5- Minerals Calcium Magnesium Phosphate Potassium 6- Blood Disorders Iron Transferrin TIBC Vitamin B12 Vitamin D Folic acid 7-Diabetes mellitus 8-Miscellaneous C-reactive protein Glycated hemoglobin (HbA1c) Uric acid 9-Cancer 10-Digestion and absorption 1- Electrolytes Sodium Potassium Chloride Bicarbonate 2- Renal (Kidney) Function Tests 6- Blood Disorders Iron Transferrin TIBC Vitamin B12 Vitamin D Folic acid 7-Diabetes mellitus Creatinine Blood urea nitrogen 3- Liver Function Tests 8-Miscellaneous Total protein (serum) Albumin Globulins A/G ratio (albumin-globulin) Protein electrophoresis Urine protein Bilirubin; direct; indirect; total Aspartate transaminase (AST) Alanine transaminase (ALT) Gamma-glutamyl transpeptidase (GGT) Alkaline phosphatase (ALP 4- Cardiac Markers 9-Cancer H-FABP (Heart-type Fatty Acid Binding Protein) Troponin Myoglobin CK-MB B-type natriuretic peptide (BNP) 5- Minerals C-reactive protein Glycated hemoglobin (HbA1c) Uric acid Calcium Magnesium Phosphate Potassium 10-Digestion and absorption