Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Paracrine signalling wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Genetic code wikipedia , lookup
Point mutation wikipedia , lookup
Gene expression wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Magnesium transporter wikipedia , lookup
Expression vector wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Biochemistry wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Interactome wikipedia , lookup
Metalloprotein wikipedia , lookup
Western blot wikipedia , lookup
Homology modeling wikipedia , lookup
Protein purification wikipedia , lookup
Two-hybrid screening wikipedia , lookup
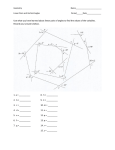
Protein Folding Pathway Prediction by Haitham Ahmad Gamal Supervised by Prof. Ibrahim M.El-Henawy Dr. Ahmed H.Kamal Dr. Hisham Al-Shishiny Problem Statement Motivation Approach Previous Work Biological Background What Affects Folding Why is it difficult Data Set Methodology (the 4 stages) Hypothesis (formally stated) Results Conclusion Proteins are the most vital agents in living bodies. Their function is what concerns scientists Function 3D Structure Hydrophobicity Much effort in structure prediction but limited success: Result are: • premature due to the huge conformations search space. • or, insufficiently accurate due to simplifications. In this study we try to limit this search space to the most likely possible conformations of a protein by answering the following questions: 1. Do angle measures depend on the hydrophobicity of the amino-acids? 2. If the answer of question (1) is "yes", how many neighbors shall be used? 3. If the answer of question (1) is "yes", what are the most likely values of the protein final structure angles? Knowledge of how a protein can fold enables us to understand how it is functioning. With this level of understanding we can affect a protein either by enhancement or by suppression. Drugs can be built to affect certain proteins directly or through other proteins interacting with the protein under investigation. The approach used in this study is a statistical, machine learning approach. We try using this approach to answer the previous questions. Clustering Distribution Fitting In our study we are not developing a prediction algorithm. Our study fits in the coloured classes across these criteria. We are proving some hypothesis that can all improve several types of prediction algorithms. Prediction algorithms/techniques can be classified based on Ab intio Homology different criteria. On-lattice Off-lattice Heuristic Statistics Protein-based Subsequence-based The tertiary structure is the minimum free energy structure of a protein (for single chain proteins) It has been proven that the function of a protein depends on its 3D structure not its primary structure. The most effective factor is folding proteins (specially globular proteins) is the hydrophobicity of its constituents amino acids. Amino acids are either charged(soluble) or contains aromatic groups(insoluble). Hydrophobicity of all the 20 known amino acids is called the Hydrophobicity scale. Residue Type Hydrophobicity Ile 4.5 Val 4.2 Leu 3.8 Phe 2.8 Cys 2.5 Met 1.9 Ala 1.8 Gly -0.4 Thr -0.7 Ser -0.8 Trp -0.9 Tyr -1.3 Pro -1.6 His -3.2 Glu -3.4 Gln -3.5 Asp -3.5 Asn -3.5 Lys -3.9 Arg -4.5 An exact simulation of a short peptide folding may take months on a super computer. The number of possible conformations is huge. such that 20l l is the length of the peptidebond Scientists proved that solving the problem for the HP model (simplified model) is NP-Complete. Current technologies cannot keep pace with this God created miracle. A collection of more than 1000 proteins is taken randomly from the SCOP protein databank Each SCOP entry (file) represents one protein with all its features including its exact atom coordinates. Angles are extracted using the three dimensional coordinates of each Cα atom Angle Extraction Chopping to Subsequences K-means Clustering Distribution Fitting X - coordinate the 3rd residue Atom Serial Number Residue Residue Name Sequence Number Y - coordinate the 4th residue Z - coordinate the 5th residue Continue doing the same until the end The angle that lies between each consecutive Cα atoms is ,called angle θ. ( , ) i-1 three Cα θ1 θ2 θ3 Let (a) be a vector such that: a = (Cαi,Cαi-1) Cαi-1 . Cα.i . . ( , , ) θ can then be calculated using the cosine law:a vector such that: b = (Cαi,Cαi+1θ) Let (b) be Cαi+1 Cαi are calculated As shown in the figure the angles Cαi+1 at each Cα atom starting ( , from, Cα1 ) until CαL-1, such that (L)is the protein length. After all the angles of all of the proteins are extracted in each protein sequence is divided into subsequences of length n. A subsequence must contain an odd number of residues. A sliding window technique is used to chop the whole protein sequence into pieces. The value of n is crucial in our study as will be shown in the results section. Let’s take n = 5 as an example aa0 aa1 aa3 Θ0 Θ2 Θ1 aa2 Θ3 aa4 aa7 aa5 Θ4 Θ7 Θ6 aa6 aa8 The first subsequence effect Similarity the effect of starts all thefrom nextaa subsequences starting 0 to aa4 and the of this subsequence on the angle Θ1 is what generally from aai to aa oncentral the measurement of the i+n-1 concerns us in this study. central angle Θ is studied. i+floor(n/2)-1 Let’s take n = 3 as an example Since hydrophobicity is the main factor affecting No. determined of proteinAll Hydrophillic folding. The centroids were initial centroids is accordingly. 2n The choice of centroids is meant to cover all the possible hydrophobicity patterns of a subsequence of length n. Hydrophobic Hydrophillic All Hydrophobic Clustered as well as the unclustered data are compared using Kolmogrov-Smirnov test against 66 continuous probability distributions, which are: Beta, Burr, Burr (4P), Cauchy, Chi-Squared, Chi-Squared (2P), Dagum, Dagum (4P), Erlang, Erlang (3P), Error, Error Function, Exponential, Exponential (2P), Fatigue Life, Fatigue Life (3P), Frechet, Frechet (3P), Gamma, Gamma (3P), Gen. Extreme Value, Gen. Gamma, Gen. Gamma (4P), Gen. Logistic, Gen. Pareto, Gumbel Max, Gumbel Min, Hypersecant, Inv. Gaussian, Inv. Gaussian (3P), Johnson SB, Johnson SU, Kumaraswamy, Laplace, Levy, Levy (2P), Log-Gamma, Log-Logistic, Log-Logistic (3P), LogPearson 3, Logistic, Lognormal, Lognormal (3P), Nakagami, Normal, Pareto, Pareto 2, Pearson 5, Pearson 5 (3P), Pearson 6, Pearson 6 (4P), Pert, Phased Bi-Exponential, Phased Bi-Weibull, Power Function, Rayleigh, Rayleigh (2P), Reciprocal, Rice, Student's t, Triangular, Uniform, Wakeby, Weibull and Weibull (3P). Through conducting this study we try to argue about two assumptions: I. The first part of the hypothesis suggests that the angles measurements of a protein sequences follow some sort of pattern based on the hydrophobicity of the surrounding amino acid residues. II. The second part suggests that the these patterns as the neighboring amino acid residues taken consideration . of of into n=3 Distribution Burr Centroids in this distribution (i = Ci) 1, 4 Burr(4p) 7 Gen. Extreme Value 6 Gen. Pareto 2, 3, 5 Johnson SB 0 n=5 Distribution Dagum(4p) Gumbel Min. Gen. Extreme Value Burr(4p) Weibull(3p) Centroids in this distribution (i = Ci) 0, 5, 7, 19 1, 2, 3, 17, 20 4, 32 6, 8, 10, 11, 14, 18, 21, 22, 23, 24, 27, 30, 31 9, 12, 13, 15, 16, 25, 26, 28, 29 n=7 Distribution Weibull(3p) Burr(4p) Dagum Dagum(4p) Gen. Gamma(4p) Gen. Logistic Gumbel Min. Log-Logistic Wakeby Centroids in this distribution (i = Ci) 3, 21, 79 20, 32, 40, 60, 67, 71, 74, 75, 83, 85, 105 4, 80 41, 90 69, 84, 106 2, 6, 7, 9, 12, 14, 15, 19, 33, 34, 35, 36, 37, 45, 46, 47, 49, 79, 87, 89, 94, 95, 107, 117, 125 66 42, 116, 118 1, 5, 8, 10, 11, 13, 16, 17, 18, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 38, 39, 43, 44, 48, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 61, 62, 63, 64, 65, 68, 70, 72, 73, 76, 77, 78, 81, 82, 86, 88, 91, 92, 93, 96, 98, 99, 100, 101, 102, 103, 104, 108, 109, 110, 111, 112, 113, 114, 115, 119, 120, 121, 122, 123, 124, 126, 127 Tricky KS-statistic value are not enough for complete interpretation KS statistic for Unclustered data KS statistic for Clustered data n=3 0.09041 0.0937 n=5 0.012 0.0243 n=7 0.013 0.0202 The number of rejected critical values shows that the fits of Un-clustered data are fake fits No. of rejected values for Un-Clustered data No. of rejected values for Clustered data n=3 All 5 values All 5 values n=5 All 5 values 2.94 n=7 All 5 values Zero Number of tested critical values is 5 Obviously the KS-statistic shows that the larger the value of n the better the fit. Looking deeper at the rejected value test, all the 5 test values are rejected for n = 3 while n = 7 gives ZERO rejected values, the thing that emphasizes the truth of our hypothesis. it is now clear that there exists a direct relationship between the hydrophobicity of the residues of a subsequence (local neighbours) and the measurements of the backbone angles. Classifying a subsequence into one of the available clusters will give a good insight of the angles measurements and consequently the structure of the subsequence. Also the length of the subsequence is an effective factor in angle measurement prediction process. Longer subsequences achieve better fits in one of the standard continuous probability distributions. These results can be used to guide the search process in a complete protein structure prediction algorithm. Local angle-hydrophobicity relationship can be used combined with heuristic techniques like genetic algorithm to restrict the initial population to statistically familiar conformation. Approximations of our results can be applied to crystalline lattices protein models like cube octahedron lattice model which allows the use of several possible angles 60", 90", 120" and 180". it is possible to investigate applying the same approach on subsequences of length more than 7 residues and try to minimize the required processing time. Title A CENTRAL-3-RESIDUES-BASED CLUSTERING APPROACH FOR STUDYING THE EFFECT OF HYDROPHOBICITY ON PROTEIN BACKBONE ANGLES Authors Prof. Ibrahim M.El-Henawy Dr. Hisham Al-Shishiny Dr. Ahmed H.Kamal Haitham Gamal Has been published in Egyptian Computer Science Journal (ECS Journal), ISSN-1110-2586, Volume 32, Number 1, May, 2009