Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Biosynthesis wikipedia , lookup
Expression vector wikipedia , lookup
Non-coding DNA wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Magnesium transporter wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Gene expression wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Genetic code wikipedia , lookup
Interactome wikipedia , lookup
Metalloprotein wikipedia , lookup
Biochemistry wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Western blot wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Structural alignment wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Proteolysis wikipedia , lookup
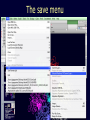
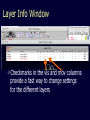
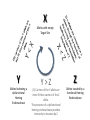
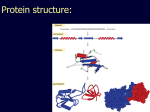
Simplify the display Show only alpha carbons Turn off show backbone oxygen Colour secondary structure Turn 3 D display on 1HEW Backbone as alpha carbons Sidechains only Tyr62 Substrate with VDW surface and CPK colors 1bmf 1bmf Colored by chain (discuss similarity to helicase and T3SS) 1bmf Open pdb – check chain id Select Chain E (by clicking in column designation in Control Panel) Safe selected residues in current layer as betaTP Repeat for betaDP, betaE, alpha TP, alphaDP, and alpha E. The save menu 1bmf Open the three beta SU <SHIFT> Color in in secondary structure (shift makes it act on the 3 layers simultaneously) 1bmf 3 beta subunits Use Magic fit and interative magic fit to align the three subunits (use betDB as reference layer) <ctrl>tab allows you to move through the three layers For an animated GIF see http://web2.uconn.edu/gogarten/F1ATPasecatcycle.htm Layer Info Window Checkmarks in the vis and mov columns provide a fast way to change settings for the different layers betaTB and betaE with RMS coloring compared to betaDP Magic fit -> fit molecules -> RMS coloring RED: Long wavelength = long distance between structures BLUE: Short wavelength = short distance between structures If you need to switch the reference layer, you can do so in the SwissModel menu The 3 point alignment tool If you want to compare the structure of very dissimilar proteins that use a similar substrate, sometimes it helps to align the substrates. This can be done through the 3 point alignment tool. Homing Homing cycle of a parasitic genetic element (modified from [3, 13]). Recent findings suggest that due to complex population structure the cycle might not operate in synchrony in different subpopulations. The red arrows indicate the trajectory of the functioning HE and the black arrows the fate of the host gene. The precise loss can occur through recombination with an intein or intron free allele, or, in case of introns, through recombination with a reverse transcript of the spliced mRNA [39, 40]. X Alleles with empty Target Site Y Alleles harboring a dysfunctional Homing Endonuclease Y>Z (II) Carriers of the Y-allele are more fit than carriers of the Z allele. The presence of a dysfunctional homing endonuclease provides immunity to invasion by Z Z Alleles invaded by a functional Homing Endonuclease Theodosius Dobzhansky "Nothing in biology makes sense except in the light of evolution" Homology by Bob Friedman bird wing bat wing human arm homology vs analogy A priori sequences could be similar due to convergent evolution Homology (shared ancestry) versus Analogy (convergent evolution) bird wing bat wing butterfly wing fly wing What does Bioinformatics have to do with Molecular Evolution? Problem: Application of first principles does not (yet) work: Most scientists believe in the principle of reductionism (plus new laws and relations emerging on each level), e.g.: DNA sequence -> transcription -> translation -> protein folding -> protein function (catalytic and other properties) -> properties of the organism(s) -> ecology At several steps along the way from DNA to function our understanding of the chemical and physical processes involved is incomplete and computational simulations are so time consuming that prediction of protein function based on only a single DNA sequence is at present impossible (at least for a protein of reasonable size). Related Proteins Present day proteins evolved through substitution and selection from ancestral proteins. Related proteins have similar sequence AND similar structure AND similar function. In the above mantra "similar function" can refer to: •identical function, •similar function, e.g.: •identical reactions catalyzed in different organisms; or •same catalytic mechanism but different substrate (malic and lactic acid dehydrogenases); •similar subunits and domains that are brought together through a (hypothetical) process called domain shuffling, e.g. nucleotide binding domains in hexokinse, myosin, HSP70, and ATPsynthases. homology Two sequences are homologous, if there existed an ancestral molecule in the past that is ancestral to both of the sequences Homology is a "yes" or "no" character (don't know is also possible). Either sequences (or characters share ancestry or they don't (like pregnancy). Molecular biologist often use homology as synonymous with similarity of percent identity. One often reads: sequence A and B are 70% homologous. To an evolutionary biologist this sounds as wrong as 70% pregnant. Sequence Similarity vs Homology The following is based on observation and not on an a priori truth: If two (complex) sequences show significant similarity in their primary sequence, they have shared ancestry, and probably similar function. (although some proteins acquired radically new functional assignments, lysozyme -> lactalbumin). The Size of Protein Sequence Space (back of the envelope calculation) Consider a protein of 600 amino acids. Assume that for every position there could be any of the twenty possible amino acid. Then the total number of possibilities is 20 choices for the first position times 20 for the second position times 20 to the third .... = 20 to the 600 = 4*10780 different proteins possible with lengths of 600 amino acids. For comparison the universe contains only about 1089 protons and has an age of about 5*1017 seconds or 5*1029 picoseconds. If every proton in the universe were a super computer that explored one possible protein sequence per picosecond, we only would have explored 5*10118 sequences, i.e. a negligible fraction of the possible sequences with length 600 (one in about 10662). no similarity vs no homology If two (complex) sequences show significant similarity in their primary sequence, they have shared ancestry, and probably similar function. THE REVERSE IS NOT TRUE: PROTEINS WITH THE SAME OR SIMILAR FUNCTION DO NOT ALWAYS SHOW SIGNIFICANT SEQUENCE SIMILARITY for one of two reasons: a) they evolved independently (e.g. different types of nucleotide binding sites); or b) they underwent so many substitution events that there is no readily detectable similarity remaining. Corollary: PROTEINS WITH SHARED ANCESTRY DO NOT ALWAYS SHOW SIGNIFICANT SIMILARITY.