Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Expression vector wikipedia , lookup
Photosynthetic reaction centre wikipedia , lookup
Magnesium transporter wikipedia , lookup
Gene expression wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Genetic code wikipedia , lookup
Interactome wikipedia , lookup
Point mutation wikipedia , lookup
Metalloprotein wikipedia , lookup
Western blot wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Biochemistry wikipedia , lookup
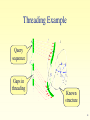
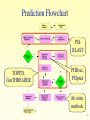
Introduction to Bioinformatics for Medical Research Gideon Greenspan [email protected] Lecture 10 Protein Tertiary (3D) Structure Protein Tertiary Structure • Defining Structure • Determining experimentally – PDB • Predicting Structure – TOPITS – GenTHREADER • Structural classification – SCOP 2 Defining Structure Atomic symbol 1 N 2 CA 3 C 4 O 5 CB 6 CG 7 SD 8 CE 9 1H 10 2H MET MET MET MET MET MET MET MET MET MET A A A A A A A A A A Hydrogen number Remoteness 1 1 1 1 1 1 1 1 1 1 Residue number -14.830 -14.608 -15.821 -15.713 -13.372 -13.531 -12.739 -13.839 -15.554 -13.942 Residue -2.121 -1.535 -1.799 -2.464 -2.254 -3.764 -4.636 -6.072 -2.865 -2.531 Chain 10.034 8.679 7.781 6.770 8.135 8.330 6.956 6.937 9.976 10.386 3D co-ords 3 X-ray Crystallography • Create repetitive crystal of molecule – Often difficult, especially hydrophobic portions • X-rays generate diffraction pattern – Pattern represents electron density • Generate comparison patterns – Add ions or change wavelength • Obtain electron density map – Fit protein sequence to map 4 Nuclear Magnetic Resonance • Dissolve molecules in water – Allows free tumbling and vibration • Detect activity of atoms with quantum spin – 1Hydrogen (natural), 13Carbon, 15Nitrogen • Defines set of atomic distance constraints – Ensemble of models • Can detect motion 5 PDB • Database of molecular structures – Obtained by crystallography or NMR – Carefully curated and validated • Founded in 1971 – 19375 proteins, 2117 other structures • Additional protein information – Secondary structure – References, external links 6 PDB: Summary Information Molecule in PDB entry Chains in molecule Experimental method Link to SCOP 7 PDB: 3D Structure • Still images at fixed orientation – Generate at any size • Interactive molecule explorer – Requires Java or Chime plug-in • Download structure file – Display in RasMol, Swiss-PDBViewer, etc… • Demonstration 8 Predicting 3D Structure • Outstanding difficult problem • Based only on protein sequence – Comparative modeling (homology) – Ab-initio modeling • Based on secondary structure – Fold recognition – Protein threading 9 Comparative Modeling • Similar sequence suggests similar structure – Amino acid characteristics determine folding • Similarity particularly high in core – Alpha helices and beta sheets preserved – But even near-identical sequences vary in loops • Effectiveness depends on protein length – Longer fi less sequence similarity required 10 Ab Initio Modeling • Compute molecular structure from laws of physics and chemistry alone – Ideal solution (theoretically) • Simulate process of protein folding – Apply minimum energy considerations • Practically nearly impossible – Exceptionally complex calculations – Biophysics understanding incomplete 11 Protein Folds • A combination of secondary structural units – Forms basic level of classification • Each protein family belongs to a fold – Estimated 700–1500 different folds 12 Fold Recognition / Threading • Compare sequence against known structures – Try to ‘thread’ sequence along chain • Score suitability of the threading – Can adjacent amino acids bond? – Are amino acids close to or far from water? – Are secondary structures similar? • Examine list of most threadable structures – Correct answer is often in top 10 or so 13 Threading Example Query sequence Gaps in threading Known structure 14 TOPITS Output (1) Alignment score Length of indels Alignment length Number of indels % sequence identity Alignment significance Matched sequence Length of sequence 15 TOPITS Output (2) Predicted structure Amino acid matches Query sequence Database sequence Buried / Outside Database known secondary structure 16 GenTHREADER Output Prediction confidence Score from neural network Energy measurements Expected errors Sequence alignment score and length Length of sequence Structure from PDB 17 Prediction Flowchart PSIBLAST TOPITS, GenTHREADER PHDsec, PSIpred Ab initio methods 18 Structure Classification • Class – All alpha, all beta, alpha/beta, alpha+beta • Fold – Strong structural similarity • Superfamily – Probably common evolutionary origin • Family – Evolutionary relationship, sequence similarity 19 SCOP • Structural Classification of Proteins – Based on known protein structures – Manually created by visual inspection • Hierarchical database structure – Class, fold, superfamily, family – Proteins/domains, species instances • Founded in 1995 – 765 folds, 1232 superfamilies, 2164 families 20 SCOP: Navigation Node name Node description Path from root to node Children of node 21 Other Resources • CATH (classification of protein domains) – http://www.biochem.ucl.ac.uk/bsm/cath/ • SWISS-MODEL (comparative modeling) – http://www.expasy.ch/swissmod/ • CASP (structure prediction competition) – http://predictioncenter.llnl.gov/ • GTSP (guide to structure prediction) – http://speedy.embl-heidelberg.de/gtsp/ 22