Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Drug design wikipedia , lookup
Magnesium transporter wikipedia , lookup
Genetic code wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Expression vector wikipedia , lookup
Biochemistry wikipedia , lookup
Gene expression wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Interactome wikipedia , lookup
Metalloprotein wikipedia , lookup
Point mutation wikipedia , lookup
Western blot wikipedia , lookup
Protein purification wikipedia , lookup
Proteolysis wikipedia , lookup
Protein–protein interaction wikipedia , lookup
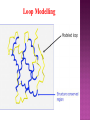
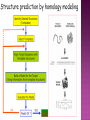
Presented by Sadhana S Protein structure prediction/protein modelling is the prediction of the three-dimensional structure of protein from its amino acid sequence i.e., the prediction of its folding & its secondary, tertiary, & quaternary structure from its primary structure Why to predict protein structure? Owing to significant efforts in genome sequencing over nearly three decades, gene sequences from many organism have been deduced. Over 100 million nucleotide sequences from over 300 thousand different organisms have been deposited in the major DNA databases, DDBJ/ EMBL/GenBank totaling almost 200 billion nucleotide bases. Over 5 million of these nucleotide sequences have been translated into amino acid sequences and deposited in the UniProtKB database. However, the protein sequences themselves are usually insufficient for determining protein function as the biological function of proteins is intrinsically linked to three dimensional protein structure. The most accurate structural characterization of proteins is provided by X-ray crystallography and NMR spectroscopy. Owing to the technical difficulties and labor intensiveness of these methods, the number of protein structures solved by experimental methods lags far behind the accumulation of protein sequences Many proteins are simply too large for NMR analysis and cannot be crystallized for X-ray diffraction. Protein modeling(computational methods) is the only way to obtain structural information if experimental techniques fail. The ultimate goal of protein modeling is to predict a structure from its sequence with an accuracy that is comparable to the best results achieved experimentally. Can we predict structure from sequence? Computational Methods The three major approaches for threedimensional (3D) structure predictions are Ab initio methods Threading methods Comparative modelling / homology modelling What is Homology Modelling? It is the prediction of the three-dimensional structure of a given protein sequence (target) based on an alignment to one or more known protein structures (templates). If similarity between the target sequence and the template sequence is detected, structural similarity can be assumed. Homology Modelling Homology modeling, also known as Comparative modeling of protein is the technique which allows to construct an unknown atomic-resolution model of the "target" protein from: 1. Its amino acid sequence and 2.An experimental 3Dstructure of a related homologous protein (the "template"). Basis for homology modelling? 1. Structure of a protein is uniquely determined by its amino acid sequence 2. Structure is much more conserved than sequence during evolution. Proteins sharing high sequence similarity should have similar protein fold. Higher the similarity, higher is the confidence in the modeled structure. Homology modeling is a multistep process that can be summarized in seven steps: 1. Template recognition & initial alignment 2. Alignment corrections 3. Backbone generation 4. Loop modeling 5. Side-chain modeling 6. Model optimization 7. Model validation TEMPLATE RECOGNITION Achieved by searching the PDB of known protein structures using the target sequence as the query. Templates can be found using the target sequence as a query for searching using FASTA or BLAST, & PSI-BLAST or PDB-BLAST Select the best template(min.30%) from a library of known protein structures derived from the PDB. ALIGNMENT Purpose – to propose the homologies between the sites in two or more sequences Insertions & deletions are placed Types 1. Pairwise alignment 2. Multiple alignment Correct alignment is necessary to create the most probable 3D structure of the target. If sequences aligns incorrectly, it will result in false positive or negative results. Important gap steps to consider: penalties Scoring alignments Alignment algorithms Alignment Corrections Alignments are scored (substitution score) in order to define similarity between 2 amino acid residues in the sequences A substitutions score is calculated for each aligned pair of letters. Alignment FASTA algorithms- DPA, BLAST & Structure of alignment 1 and 2 with the template Alignment Outcome The (true) alignment indicates the evolutionary process giving rise to the different sequences starting from the same ancestor sequence and then changing through mutations (insertions, deletions, and substitutions) BACKBONE GENERATION One simply copies the coordinates of those template residues that show up in the alignment with the model sequence If two aligned residues differ- only backbone coordinates(N, C-alpha, C & O) are copied It they are same- side chain is also included Backbone Generation For SCRs - copy coordinates from known structures. For variable regions (VR) - copy from known structure, if the residue types are similar; otherwise, use databases for loop sequences. Loop Modelling Knowledge based- PDB is searched 2. Energy based- energy function is used to judge the quality of loop Molecular modeling/dynamic programs are used 1. Loop Modelling Side Chain Modelling 1. Use of rotamer libraries (backbone dependent) 2. Molecular mechanics optimization - Dead-end elimination (heuristic) - Monte Carlo (heuristic) - Branch & Bound (exact) Model refinement/optimization Idealization of bond geometry Removal of unfavorable non-bonded contacts Performed by energy minimization with force fields such as CHARMM, AMBER, or GROMOS Major errors are removed Evaluation/validation of the model Internal evaluation Self-consistency checks Assessment of stereochemistry of the model PROCHECK & WHATCHECK External evaluation Tests whether a correct template was used PROSA & VERIFY3D Applications Designing mutants to test hypotheses about the function of a protein. Identifying active & binding sites. Predicting antigenic epitopes. Simulating protein-protein docking. Confirming a remote structural relationship. Web servers Swiss- model server (http://www.expasy.ch/swissmod/) CPHModels (http://www.cbs.dtu.dk/services/CPHmodels/) SDSC1 (http://www.cl.sdsc.edu/hm) FAMS (http://www.physchem.pharm.kitasatou.ac.jp/FAMS/fams.html) ModWeb (http://www.guitar.rockefeller.edu/modweb) References Zhumur Ghosh & Bibekanand mallik. bioinformaticsPrinciples & applications. Oxford university press S C Rastogi, N.Mendiratta, & P Rastogi. Bioinformaticsmethods & applications. Eastern economy edition. Prentice hall of India. New Delhi Philip.E.Bourne & Helge Wiessig. Structural Bioinformatics. John Wiley & Sons. NewYork C A Orengo, D T Jones & J M Thornton. Bioinformaticsgene, proteins, & computers. BIOS . Scientific Publishers