Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gene expression wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Expression vector wikipedia , lookup
Magnesium transporter wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Peptide synthesis wikipedia , lookup
Point mutation wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Genetic code wikipedia , lookup
Biosynthesis wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Homology modeling wikipedia , lookup
Protein purification wikipedia , lookup
Western blot wikipedia , lookup
Metalloprotein wikipedia , lookup
Interactome wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Biochemistry wikipedia , lookup
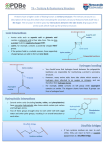
7/14/2009 ` ` ` ` ` ` ` ` Proteins are polymers consisting of amino acids linked by peptide bonds Proteins contain a wide range of functional groups Proteins P i can interact i with i h one another h and d with ih other biological molecule The function of a protein depend on its 3dimensional shape The amino acid sequence of a protein determines the 3D shape of a protein Each amino acid consist of • a central carbon atom • an amino group, NH2 • a carboxyl group, COOH • a side chain Amino acid side chains vary in shape, size, polarity and charge http://www.ecosci.jp/amino/amino2j_e.html 1 7/14/2009 Aliphatic Aromatic Nonpolar Polar Charged Ala Tyr Gly Asn Asp Val Trp Cys Gln Glu Leu Phe Pro Ser Lys Ile His Met Thr Arg 2 7/14/2009 ` Resonance structure: ` The properties of the peptide bond have important effects on the stability and flexibility of polypeptide chains in water B Because off resonance, ◦ the polarity of peptide bond increases. ◦ the peptide bond has partial double bond character; carbonyl carbon, Cα, and amide N are coplanar and free rotation about the bond is restricted. 3 7/14/2009 ` ` Which configuration is more stable? ` ` ` ` Used computer models of small polypeptides to systematically vary φ and ψ with the objective of finding stable conformations Atoms were treated as hard spheres with dimensions corresponding to their van der Waals radii For each conformation, the structure was examined for close contacts between atoms Therefore, φ and ψ angles which cause spheres to collide correspond to sterically disallowed conformations of the polypeptide backbone The angle of the N-Cα bond to the adjacent peptide bond is known as phi (φ) torsion angle and the angle of the C-Cα bond to the adjacent peptide bond is known as psi (ψ) Physical size of atoms or group of atoms limits the possible ψ and φ angles ` ` Plot of φ vs. ψ The computed angles which are sterically allowed fall on certain regions of plot White h = sterically ll disallowed d ll d conformations (atoms come closer than sum of van der Waals radii) Blue = sterically allowed conformations 4 7/14/2009 φ, ψ distribution in 42 high-resolution protein structures (x-ray crystallography) Ramachandran Plot and Secondary Structure The structure of cytochrome C shows many segments of helix and the Ramachandran plot shows a tight grouping of φ, ψ angles near -50,-50 alpha-helix Ramachandran plot: Cytochrome C Ramachandran Plot The structure of plastocyanin is composed mostly of beta sheets; the Ramachandran plot shows values in the –110, +130 region: beta-sheet ¾Protein structure has a hierarchical organization. Ramachandran plot: Plastocyanin 5 7/14/2009 ` ` Rigidity of backbone ` Interactions among amino acids ` Interactions of amino acids with water Covalent -Cα-C- Disulphide ` Non-covalent Electrostatic interaction (Salt bridge and long-range) Hydrogen bonding van der Waals interactions (transient, weak electrical attraction of one atom for another) Hydrophobic (clustering of nonpolar groups) 6 7/14/2009 ` ¾ ¾ Side chain of cysteine contains highly reactive thiol group; two thiol groups form a disulfide bond Contribute to the stability of the folded state by linking distant parts of the polypeptide chain Typical distance 2.2 Å and contribute ~200 KJ/mol PDB: 5PTI Charge-charge Interactions Charge(The “Salt bridge” Interactions) q1 NH3 ` Charged side chains in protein can interact favorably with an opposing charge of another side chain according to Coulomb’s law. q2 r O O ` C k q1q2 Force = ε r2 ` Examples of favorable electrostatic interaction include that between positively charged lysine and negatively charged glutamic acid. In practice, charge-charge interactions have been shown to be chemically significant at up to 15 Å in proteins. ` Contribute less than 5 kJmol-1 to the overall stability of a protein. ` Salts have the ability to shield electrostatic interactions. 7 7/14/2009 (Adopted from Garrett and Grisham, Biochemistry, Third Edition) Hydrogen Bonds N DONOR H H Hydrogen Bonds O=C ACCEPTOR CC O δ+ δ- δ- H-bond length (fixed for a given donor-acceptor pair) 8 7/14/2009 ` ` Most common type of 2˚ structural element (in globular proteins, over 30% of the amino acids are found in helices) ` Generated by local hydrogen bonding between C=O and N-H groups (intrachain hydrogen bonding) ` R-groups project outward, and provide the main constraints on helical structure ` Stability is greatly enhanced by internal van der Waals contacts ` Each residue is related to the next one by a translation 1.5 Å along the helix axis and rotation of 100 degree ` 3.6 residues per turn of a helix and pitch of 5.4 Å ` Right-handed helix are energetically more favorable Involves hydrogen bonds between backbone groups from residues distant from each other in the linear sequence Antiparallel β sheet Parallel β sheet Two residues per turn and a translation distance of 3.4 Å, pitch of 6.8 Å 9 7/14/2009 y The simplest secondary structure elements is the beta turn: H-bond between the carbonyl oxygen of one residues (n) and the amide N-H of residue n = 3, reversing the direction of the chain y β turn-four residues and γ turn- three residues Hydrogen Bonds ` ` Hydrogen bonds between main chain NH and CO groups Figure Hydrogen bonds between i. side chains ii. side chain - main chain Side chains of tyrosine, threonine, and serine containing hydroxyl group the side chains of glutamine and asparagine with the amide group 10 7/14/2009 Hydrogen Bonds ` ` ` ` These interactions occur between adjacent, non-bonded, and uncharged atoms. • Dipole-dipole interactions • Dipole-induced dipole interactions ` ` What type of forces must be overcome within the solid I2 when I2 dissolves in methanol, CH3OH? What type of forces must be disrupted between CH3OH molecules when I2 dissolves? What type of force exists between I2 and CH3OH molecules in solution? van der Waals interaction diminish rapidly as the interacting species get farther apart; atoms that are about 5 Å apart can have significant interactions. A given van der waals interaction is extremely weak 4 kJmol-1 but in proteins they sum up to a substantial energetic contribution. • Induced dipole-induced dipole interactions (London dispersion forces, LDF) 11 7/14/2009 ¾ Observation: • hydrophobic groups aggregate together on the interior of the protein, forming a hydrophobic core while hydrophilic residues are on the outside. • The exterior surface area of proteins can be up to 60% polar atoms • Proteins fold to maximize their effectiveness as hydrogen-bonding partners to water ¾ Water molecules surrounding the nonpolar molecules are more ordered than water molecules in free solution The aggregation of nonpolar groups in water leads to the release of water molecules Explanation: • When hydrophobic residues are exposed to solvent, the extended hydrogen bonding structure of water is disrupted • Breaking hydrogen bonds in water is energetically unfavourable • Water molecules reorient around the hydrophobic molecule, so that the least number of hydrogen bonds are sacrificed to accommodate it • Burying hydrophobic residues releases water and increases entropy. Approximate strengths of interactions between atoms ` ` Interaction Typical distance Energy Salt bridge 2.8 Å 12.5 – 17 kJ/mol Long-range electrostatic interactions variable Variable, depends upon the environment Hydrogen bond 2.4 – 3.5 Å 2- 6 kJ/mol van der Waals interactions 3.5 – 6.0 Å 2-4 kJ/mol ` ` ` Secondary structures pack closely to one another and also intercalate with extended polypeptide chains Most polar residues face the outside of protein and interact with solvent Most hydrophobic residues face the interior of the protein and interact with each other thereby minimizing contact with water van der Waal’s volume is about 72-77% of the total protein volume; about 25% is not occupied by protein atoms. These cavities provide flexibility in protein conformation and dynamics Random coil or loops maybe of importance in protein function (interacting with other molecules, enzyme reactions) 12 7/14/2009 ` Water soluble proteins fold into compact structure with nonpolar cores ` Membrane proteins fold into compact structure with polar cores charged hydrophobic ` Folding of a globular protein is a thermodynamically Change in Entropy and enthalpy in protein folding favored process, i.e. ΔG must be negative. ΔG = ΔH - TΔS ` The folding process involves going from a multitude of random-coil conformations to a single folded structure. ` The folding process involves a decrease in randomness and thus a decrease in entropy -ΔS and an overall positive contribution to ΔG. This decrease in entropy is termed “conformational entropy”. ` An overall negative ΔG : a result of features that yield a large negative ΔH or some other increase in entropy on folding. Folded Protein Unfolded Protein ΔH, large; ΔS, small ΔH, small; ΔS, large ΔG = ΔH - TΔS ΔSsurr = − ΔH sys T 13 7/14/2009 ΔG, Gibbs Free Energy Transition state, energy barrier ΔG U Unfolded f ld d State Native State Reaction Coordinate Anfinsen’s Experiments Protein Folding “The native, folded structure of a protein, under optimal conditions, is the most energetically stable conformation possible” Christian Anfinsen, 1972 “Most of the information for determining the threedimensional structure of a protein is carried in its amino acid sequence” Anfinsen, C.B. Principles that govern the folding of protein chains. Science 181, 223-30 (1973). Anfinsen’s Experiments: Unfolding of Ribonuclease ¾Ribonuclease: Involved in cleavage of nucleic acids , structure has a combination of α and β segments, four disulfide bridges ¾Unfolding and refolding experiments were conducted 14 7/14/2009 Anfinsen’s Experiments Reduction of Disulfide Bonds Dialysis β-mercaptoethanol ¾Observation: Enzyme spontaneously folded into a catalytically active form ¾Conclusion: The information need to specify the catalytically active structure of ribonuclease is contained in its primary sequence ` Protein Folding is a highly Cooperative Process “all or none” process 15 7/14/2009 ` ` ` For any given protein, there is one conformation that has significantly lower free energy than any other state Achieved through kinetic pathway of unstable intermediates (not all intermediates are sampled) ` ` The hydrophobic effect –the clustering of hydrophobic side chains from diverse parts of the protein chain- causes the protein chain to fold into compact, ordered form. Why this effect is thermodynamically favored? Entropy-driven process Assisted by chaperones and protein disulfide isomerases Consider a protein of 100 amino acids. Assign 2 conformations to each amino acid. The total conformations of the protein is 2100=1.27x1030. Allow 10-13 sec for the protein to sample through one conformation in search for the overall energy minimum. The time it needs to sample through all conformations is (10-13)(1.27x1030)=1.27x1017sec = 4x109 years! Levinthal’s paradox illustrates that proteins must only sample through limited conformations, or fold by “specific pathways”. Much research efforts are devoted in searching for the principles of the “specific pathways”. 16