Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Marcus theory wikipedia , lookup
Discodermolide wikipedia , lookup
Cracking (chemistry) wikipedia , lookup
Homoaromaticity wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
George S. Hammond wikipedia , lookup
2-Norbornyl cation wikipedia , lookup
Asymmetric induction wikipedia , lookup
Asymmetric hydrogenation wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Bottromycin wikipedia , lookup
Petasis reaction wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Metal carbonyl wikipedia , lookup
Enantioselective synthesis wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
The University of Toledo
The University of Toledo Digital Repository
Theses and Dissertations
2013
Investigation of the steric and electronic properties
of 3-iminophosphine ligands in chelated palladium
allyl complexes for use in the regioselective
hydroamination of allenes
Nicholas Charles Zingales
The University of Toledo
Follow this and additional works at: http://utdr.utoledo.edu/theses-dissertations
Recommended Citation
Zingales, Nicholas Charles, "Investigation of the steric and electronic properties of 3-iminophosphine ligands in chelated palladium
allyl complexes for use in the regioselective hydroamination of allenes" (2013). Theses and Dissertations. 248.
http://utdr.utoledo.edu/theses-dissertations/248
This Thesis is brought to you for free and open access by The University of Toledo Digital Repository. It has been accepted for inclusion in Theses and
Dissertations by an authorized administrator of The University of Toledo Digital Repository. For more information, please see the repository's About
page.
A Thesis
entitled
Investigation of the Steric and Electronic Properties of 3-Iminophosphine Ligands in
Chelated Palladium Allyl Complexes for Use in the Regioselective Hydroamination of
Allenes
by
Nicholas Charles Zingales
Submitted to the Graduate Faculty as partial fulfillment of the requirements for the
Master of Science Degree in Chemistry
____________________________________
Dr. Joseph A.R. Schmidt, Committee Chair
____________________________________
Dr. Mark R. Mason, Committee Member
____________________________________
Dr. Jianglong Zhu, Committee Member
____________________________________
Dr. Patricia R. Komuniecki, Dean
College of Graduate Studies
The University of Toledo
May 2013
Copyright 2013, Nicholas Charles Zingales
This document is copyrighted material. Under copyright law, no parts of this document
may be reproduced without the expressed permission of the author.
An abstract of
Investigation of the Steric and Electronic Properties of 3-Iminophosphine Ligands in
Chelated Palladium Allyl Complexes for Use in the Regioselective Hydroamination of
Allenes
by
Nicholas Charles Zingales
Submitted to the Graduate Faculty as partial fulfillment of the requirements for the
Master of Science Degree in Chemistry
The University of Toledo
December 2012
A series of (3-iminophosphine)allylpalladium triflate complexes varying in both
steric and electronic features was isolated and characterized. The propensity of the
complexes in this series to regioselectively catalyze the hydroamination of 3-methyl-1,2butadiene with aryl amines to form solely the kinetic product at room temperature was
studied by observing conversion to products via NMR spectroscopy. An understanding of
the importance of these 3-iminophosphine ligand features guides the rational design of
future catalysts. The 3-iminophosphine ligand composed of di-tert-butyl phosphine,
cyclobutene backbone, and tert-butyl imine provided the most active palladium catalyst
for this hydroamination when compared to the other complexes in the collection. Larger
alicyclic backbone sizes decrease catalytic activity, as does steric hindrance at the imine
substituent group. Tertiary aliphatic groups on the phosphorus are necessary for effective
catalysis, as primary and secondary aliphatics and aromatic groups also decrease the
catalytic activity. Highly basic imine moieties were also shown to decrease catalytic
activity.
iii
Acknowledgments
I would like to start of by acknowledging my parents and brother. Without your
love and support over the past couple of decades, I don’t think I’d be half the person that
I am today. The past three years here while obtaining this degree have helped me to
realize all of that. To my folks, I couldn’t have asked for a better upbringing; you taught
me as much as you could and then some. Whenever I need advice you are and always
have been the first people I think to go to. To Tony, I couldn’t think of a better person to
share a room with for those 19 years growing up at home. You work harder than every
person I know, including myself, and for this I hope that you succeed in whatever career
you end up with at the end of this year. Thank you to all the members of my lab group,
especially John, for helping me get through all of the hurtles of my masters, whether it be
choosing the best methodology for a reaction or formatting a citation for the tenth time.
Thank you to my advisor, Joe, for getting the best out of me that he could and for all the
things that he taught me over the last few years. I would also like to thank the University
of Toledo for giving me the opportunity to pursue my education here, along with my
alma mater John Carroll University for all that its teachers and students taught me over
the course of my undergraduate education. Finally I would like to thank the National
Science Foundation for funding this research under CHE-0841611.
iv
Table of Contents
Abstract
iii
Acknowledgments
iv
Contents
v
List of Tables
vii
List of Figures
viii
List of Schemes
ix
Chapter 1
A Brief Introduction to Late Transition Metal Catalyzed
Hydroamination with an Emphasis on 3-Iminophosphine-Based
Palladium Complexes
Chapter 2
1.1 The Hydroamination Reaction . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1
1.2 Proposed Catalytic Mechanisms of Hydroamination . . . . . . . . . . .
2
1.3 Late Transition Metal-Catalyzed Hydroamination . . . . . . . . . . . . .
4
1.4 3-Iminophosphine Based Palladium Hydroamination Catalysts . .
9
Investigation of Steric and Electronic Features of 3Iminophosphine-Based Palladium Hydroamination Catalysts
2.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
18
2.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
31
v
2.4 Experimental Section . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
32
2.4.1 General Methods and Instrumentation . . . . . . . . . . . . . . . 32
2.4.2 General Procedure for the Catalytic Hydroamination
34
Screening of Compounds 1Pd-10Pd . . . . . . . . . . . . . . . . . . . .
2.4.3 Synthesis and Characterization of Reaction Products . . .
34
2.5 Crystallography of 1Pd, 8Pd, and 9Pd . . . . . . . . . . . . . . . . . . . . . . 47
Chapter 3
Further Modification of the 3-Iminophosphine-Based Palladium
Catalyst
3.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
3.2 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
53
3.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
59
3.4 Experimental Section . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
59
3.4.1 General Methods and Instrumentation . . . . . . . . . . . . . . . 59
3.4.2 General Procedure for the Catalytic Hydroamination
Screening of Compounds 11Pd-14Pd . . . . . . . . . . . . . . . . . . .
61
3.4.3 Synthesis and Characterization of Reaction Products . . .
62
3.5 Crystallography of 4AgCl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
68
71
References
vi
List of Tables
2.1
Comparison between selected 31P and 1H NMR resonances of
[(3IP)Pd(allyl)]OTf complexes and their respective free 3IP ligands . . . . . . . 21
2.2
Conversion to product A for the [(3IP)Pd(allyl)]OTf catalyzed
hydroamination of 1,1-dimethylallene with aryl amines (complexes 1Pd10Pd) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
29
2.3
Crystallographic data for compounds 1Pd, 8Pd, and 9Pd . . . . . . . . . . . . . . .
50
3.1
Comparison between selected 31P and 1H NMR resonances of
[(3IP)Pd(allyl)]OTf complexes and their respective free 3IP ligands . . . . . . . 56
3.2
Conversion to product A for the [(3IP)Pd(allyl)]OTf catalyzed
hydroamination of 1,1-dimethylallene with aryl amines (complexes 11Pd-
3.3
14Pd) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
58
Crystallographic data for compound 4AgCl . . . . . . . . . . . . . . . . . . . . . . . . . .
70
vii
List of Figures
2-1 3-Iminophosphine (3IP) ligands 1-10 . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
17
2-2 ORTEP diagram of 1Pd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
24
2-3 ORTEP diagram of 8Pd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
25
2-4 ORTEP diagram of 9Pd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
26
2-5 ORTEP diagrams of cis and trans isomers of 9Pd . . . . . . . . . . . . . . . . . . .
27
3-1 3-Iminophosphine (3IP) ligands 11-14 . . . . . . . . . . . . . . . . . . . . . . . . . . . .
51
3-2 ORTEP of 4AgCl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
54
viii
List of Schemes
1-1
Metal catalyzed hydroamination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4
1-2
Late-transition metal catalyzed hydroamination of substituted allenes . . . .
6
1-3
[(3-Iminophosphine)Pd(allyl)]OTf catalyzed hydroamination . . . . . . . . . .
10
1-4
Proposed catalytic cycle for the (3IP)allylpalladium triflate catalyzed
hydroamination of 1,1-dimethylallene with primary amines . . . . . . . . . . . .
2-1
11
General synthesis of [(3IP)Pd(allyl)]OTf complexes . . . . . . . . . . . . . . . . . . 19
ix
Chapter 1
A Brief Introduction to Late Transition
Metal Catalyzed Hydroamination with an
Emphasis on 3-Iminophosphine-Based
Palladium Complexes
1.1 The Hydroamination Reaction
Hydroamination is an advantageous method for the preparation of primary,
secondary, and tertiary amines due to its 100% atom economy1 and the wide range of
regio-, stereo-, and enantioselective metal complexes that are available to catalyze this
transformation.2-10 Hydroamination is the addition of an N-H bond of ammonia or of a
primary or secondary amine across an unsaturated C-C bond of an alkene, alkyne, or
allene. The reaction can take place either intermolecularly at a high entropic demand
using affordable and readily obtained substrates or intramolecularly with low entropic
demand, but requiring less accessible starting materials. Although hydroamination is
slightly exothermic, the direct [2+2] cycloaddition of an N-H bond across a C-C
unsaturated bond is orbitally forbidden under thermal conditions, making a catalytic route
for this process a virtual necessity.2
1
The pervasive nature of carbon-nitrogen bonds in pharmaceuticals and natural
products lends credence to the investigation of new methods of carbon-nitrogen bond
formation. Diversification of the hydroamination reaction’s substrate scope will provide
key tools for organic chemists to make C-N bond containing products with greater ease
and variety, allowing for the use of less reagents, time, and energy, therefore effecting a
cost savings in these processes. Similar reactions, such as allylic amination, form small
molecule byproducts that decrease their atom economy compared to that of
hydroamination.11 Due to their robustness in the presence of oxygen and moisture, as
well as their unique reactivity to catalyze the formation of certain products more readily
than other metal centers, late transition metal (TM) catalysts have become a significant
focus in the field. This product selectivity granted by late transition metal centered
catalysts is most likely due to their unique mechanistic nature compared to early and
lanthanide metal centered complexes.
1.2 Proposed Catalytic Mechanisms of Hydroamination
Late transition metal catalyzed hydroamination has two general postulated
mechanisms. One involves the nucleophilic attack of an amine upon the double bond of
an η2-alkene or an η3-allyl coordinated to a metal center. The Lewis acidic metal draws
electron density away from the double bond, increasing the susceptibility of the bound
species to nucleophilic attack (Scheme 1-1 A).2,12,13 The other mechanism involves the
coordination of the unsaturated C-C moiety and its insertion into a metal hydride bond,
followed by oxidative addition of the amine to the metal center. The newly formed amido
2
and alkyl ligands then undergo reductive elimination, releasing the hydroamination
product and reforming the active catalyst (Scheme 1-1 B).2,14,15
Early transition metal hydroamination catalysts typically exist as metal-imido
complexes that allow for the [2+2] cycloaddition of an unsaturated C-C bond to an M-N
bond to form an azametallacyclobutane ring.2,16,17 After insertion, protolytic cleavage of
the M-C bond occurs via one equivalent of amine, followed by a second protolytic
cleavage, this time of the M-N bond, releasing the product and reforming the metal-imido
catalyst (Scheme 1-1 C). The catalytic mechanism of early transition metal complexes
restricts the substrate scope of many early metal centered complexes to primary amines,
as secondary amines cannot form the metal-imido intermediate. An alternative
mechanism is available for the transformation of secondary amines via early transition
metal centered catalysts, though complexes utilizing it are far less common than those
that go through the azametallacyclobutane intermediate.
Formation of the
azametallocyclobutane ring also has a strong effect on the regiochemistry of the nitrogen
moiety’s addition; when hydroaminating allenes the nitrogen typically adds to the center
carbon of the allene to form an enamine that subsequently rearranges to form an
imine,16,18 whereas late transition metal catalysts are able to add the nitrogen to the
terminal carbons to maintain the amine and allyl functionalities.3,19-22
Rare-earth metal catalyzed hydroamination is typically limited to the
intramolecular variety, but exceptions to this trend are known.2,23 The proposed pathway
for this transformation involves the insertion of an unsaturated C-C bond into a metalamido bond, with subsequent protolytic cleavage of the hydroamination product via an
equivalent of amine to reform the active catalyst (Scheme 1-1 D).24
3
R"
R
R'
N
MLn
R'
R
H2NR"
LnM=NR"
R
R'
R"
NHR"
MLn
R'
N
NHR"
C
R
Scheme 1-1 Metal catalyzed hydroamination.
Late transition metal catalyzed hydroamination: Nucleophilic attack A.
Late transition metal catalyzed hydroamination: M-H bond insertion B.
Early transition metal catalyzed hydroamination: [2+2] Cycloaddition C.
Rare-Earth metal catalyzed hydroamination: M-N bond insertion D.
Protolytic cleavage of the M-C bond follows to release the hydroamination product, with
coordination of the newly formed amido ligand to reform the active catalyst (Scheme 1-1
D). Group (II) and group (XIII) hydroamination catalysts are proposed to proceed
through an analogous mechanism to that of the rare-earth metal centered complexes.25,26
1.3 Late Transition Metal-Catalyzed Hydroamination
There are numerous late transition metal centered systems for both inter- and
intramolecular hydroamination,2,27,28 and because of the favorable properties of these
complexes, they will be the focus of further discussion. This group of metals has shown
extremely versatile reactivity for catalyzing the hydroamination of a variety of
unsaturated
C-C
bonds
including
4
alkenes,14,15,29-40
alkynes,28,41-56
and
dienes5,7,12,14,21,22,27,30,32,33,57-71
with a variety of nitrogen containing compounds.
Expansion of the substrate scope of the hydroamination reaction is the main driving force
of the continued research into the development of new catalysts. Despite a continually
developing library of complexes that each catalyze the reaction with a very specific set of
substrates, there still remains a need for a general methodology for hydroamination
catalysis.
Activated dienes (such as conjugated 1,3-dienes and allenes) and alkynes are
more readily hydroaminated than alkenes and unactivated dienes, which is reflected in
the number of precatalysts published for the catalysis of each of these respective
transformations. At first glance, the greater electron density of the sp hybridized carbon
of an alkyne may make it seem to be less likely to interact with the electron rich nitrogen
moiety during hydroamination than the sp2 hybridized carbon of an alkene, but this is
clearly not the case. The negative change in enthalpy (ΔH) is the thermodynamic driving
force in all intermolecular hydroamination reactions while retaining an important role in
the intramolecular variety of the transformation as well. This change in enthalpy is
dependent upon the difference in the heat of formation of the hydroamination products
and its reactants, and is greater in magnitude for the transformation of an alkyne to a
substituted alkene or imine than it is for the conversion of an alkene to an alkyl
amine.72,73 The negative entropy value for intermolecular hydroamination, along with this
negative ΔH value (denoting an exothermic reaction), necessitates the use of lower
temperatures for effective hydroamination reactions.74
Due to their conjugation and the resonance stabilization of hydroamination
intermediates, 1,3-dienes and vinylarenes undergo hydroamination much more readily
5
than other dienes, whose unconjugated pi systems react much more similarly to normal
alkenes.2 Nájera,65 Yeh,7 and Toste64 have all recently reported advances in the
hydroamination of 1,3-dienes. Nájera showed that a variety of Lewis and Brønsted acids
could catalyze intramolecular hydroamination of amines tethered to 1,3-dienes in good to
excellent yields with good E:Z stereoselectivity. Yeh used a (Ph 3 P)AuCl catalyst to
intramolecularly hydroaminate cyclic 1,3-dienes, while Toste noted improved
intramolecular hydroamination enantioselectivity using the chiral alcohol menthol as an
additive.
Allene hydroamination has grown greatly in popularity over the past few years.
Although the intramolecular hydroamination of allenes has been studied briefly,59,60,62,7578
with a focus on enantioselective conversion to products, examples of intermolecular
allene hydroamination are more prevalent and include those published by Bertrand,5,21,57
Widenhoefer,20,79,80 Yamamoto,22,58 Toste,12 and Schafer,18,81 as well as our own group.3
Late transition metal catalyzed intermolecular hydroamination of substituted allenes
yields two possible regioisomers. Addition of the amino moiety to the substituted carbon
terminus of a mono-substituted allene yields a new chiral carbon center and a vinylterminated product, which is typically the kinetic product of these reactions (Scheme 12).
Scheme 1-2 Late-transition metal catalyzed hydroamination of substituted allenes. 6
Addition of the amine at the less-substituted carbon atom yields an internal alkene,
commonly the thermodynamic product (Scheme 1-2). Regioselective hydroamination of
substituted allenes to form the thermodynamic product is much more common and has
been accomplished previously with aryl amines and a gold catalyst,20,58 secondary
alkylamines and gold21,22 or platinum79 catalysts, ammonia and a gold catalyst,57 and
hydrazine with a gold catalyst.5 Also notable is Toste’s mechanistic study involving the
hydroamination of a symmetrical allene with methyl carbazate, which demonstrated
activation of the allene as the rate-limiting step and implied that there was no coordinated
amine in the catalytic mechanism.12 Regioselective intermolecular hydroamination of
substituted allenes to form the kinetic (branched) product, though much rarer, has been
accomplished via a gold(I) N-heterocyclic carbene complex4 and a rhodium(I)
cyclooctadiene chloro dimer.19 Substrate scope was limited to the addition of an Nunsubstituted carbamate in the first example, but the second example was capable of
hydroaminating mono-substituted allenes with an array of substituted anilines to the more
substituted side of the allene with good enantiomeric excess.4
Alkynes used in hydroamination are typically of the terminal variety,2 affording
the Markovnikov selectivity product in good yields with a broad range of amines,
including aryl amines,43,44,48-50,82 sulfonamides,45 and alkylamines.44 Hydroamination of
internal alkynes, though less common in practice, has regioselectivity that is usually
governed by sterics, with the nitrogen moiety adding to the least sterically hindered sp
carbon.2,47 Recent exceptions to this trend include work from Zhu, who demonstrated the
addition of aniline to the phenyl side of methyl phenylpropiolate,41 while Corma has
produced a mixture of Markovnikov and anti-Markovnikov products utilizing both
7
alkylamines and aryl amines, with product selectivity depending upon electronic rather
than steric effects.44
Alkenes with high angular strain, such as bicyclic systems like norbornene, react
more readily than unstrained pi systems, as the addition of the N-H bond and breakage of
the strained double bond create a more relaxed system in the reaction products. Recent
examples of norbornene as a substrate for late transition metal catalyzed hydroamination
include work done by Tilley83 and Hartwig,15,32 utilizing platinum and iridium catalysts,
respectively. Earlier examples of this reaction include those from Brunet84 and Beller,85
each using rhodium to catalyze the reaction.
Unstrained alkenes, such as ethylene, have a much less significant thermodynamic
driving force towards products when their pi bonds are broken compared to alkynes or
bicyclic alkene systems. Very few examples of ethylene hydroamination are present in
the literature,2 with the work of Poli and coworkers as a notable exception.86 Poli found
that aniline could be added to ethylene with increased product conversions than those
previously reported by others34,87 by utilizing aqueous bromide ion to promote the PtBr 2
catalyzed reaction. It was proposed that the bromide anion displaces amine from
coordination sites on the metal center, giving the amine more opportunity to attack the
ethylene as a nucleophile.34 There are no examples to date that display adequate
conversions to hydroaminated ethylene products; amine substrates tend to displace the
weakly coordinating ethylene ligand, severely hindering catalysis in all cases.2
Substituted alkenes have found more utility in hydroamination than ethylene, with
examples of regio-, stereo-, and enantioselective complexes being used to catalyze their
reaction.2,8,34 Cyclic and monosubstituted alkenes are more facilely transformed than
8
heavily substituted, sterically hindered alkenes.2,30,33,34,37 Intramolecular hydroamination
of aminoalkenes is more common than the intermolecular transformation and has been
shown to provide synthetic routes to a variety of nitrogen containing heterocycles.2
The type of amine used during hydroamination has a significant effect on the
success of the reaction. The steric and electronic properties of the amine used determine
its readiness to add across the carbon-carbon multiple bond. Highly basic amines, while
being predicted to act as good nucleophiles for attack on the unsaturated C-C bond, also
tend to irreversibly coordinate to the metal center, competing for precious coordination
sites necessary for alkene substrate binding and activation. This almost always results in
the “death” of the catalyst, making highly basic amines some of the most difficult
substrates to use in hydroamination.2 Amines of less basicity are unlikely to poison the
catalyst, but are also less likely to attack as nucleophiles or to coordinate to the metal
center in order to participate in further chemical transformations, such as intramolecular
proton transfer. Sterically hindered amines govern regioselectivity in many cases, such as
with the hydroamination of internal alkenes and alkynes, adding the nitrogen moiety to
the least sterically hindered carbon. Steric hindrance can also prevent reaction with many
substrates and complexes due to inaccessibility to either coordination sites on the metal
center or to the unsaturated carbons under nucleophilic attack.
1.4 3-Iminophosphine-Based Palladium Hydroamination Catalysts
To extend upon research in this field and to develop a broader hydroamination
substrate scope, the Schmidt Group has contributed multiple 3-iminophosphine (3IP)
9
palladium allyl triflate complexes capable of catalyzing a diverse range of
hydroamination reactions (Scheme 1-3).
HNRR'
HNRR'
Ph
2Pd
2Pd
NRR'
Ph
NR'
or
Ph
(R = H)
(R = H)
HNRR'
C
HNRR'
C
H2NR
NRR'
5Pd
NRR'
5Pd
NRR'
4Pd
HRN
Scheme 1-3 [(3-Iminophosphine)Pd(allyl)]OTf catalyzed hydroamination.
The
first
generation
complex,
[(2-diphenylphosphinocyclopentene-1-(tert-
butyl)imine)Pd(allyl)]OTf (2Pd), was shown to hydroaminate cyclohexadiene and phenyl
acetylene with amines of varying pK b values in moderate to good yields.88 These
hydroamination products and their yields were not exceptional, but this was the first time
that a 3-iminophosphine complex was employed in the catalysis of hydroamination,
proving that these new complexes were indeed worthy of further research pursuits. The
correlation between amine basicity and hydroamination product yield combined with the
10
lack of acid assisted catalysis also allowed the postulation of an alternative catalytic
mechanism to the common unsaturated bond activation and nucleophilic attack of amine
pathway proposed by others.2,88
The mechanism proposed by the Schmidt group is summarized in Scheme 1-4
below.
Scheme 1-4 Proposed catalytic cycle for the (3IP)allylpalladium triflate catalyzed
hydroamination of 1,1-dimethylallene with primary amines. After reductive elimination of the first turnover product of allene (from hydroamination
of the allyl ligand), the active catalyst is thought to be a palladium(II) hydride. At this
point the allene or diene coordinates to the metal center where it can readily insert into
the palladium-hydride bond. The insertion reaction is followed be either an associative or
dissociative amine coordination/imine decoordination step. This most likely happens after
diene insertion in order to release ring strain of the chelate ring. The imine moiety of the
ligand is now rendered more basic, while the amino ligand is now rendered more acidic,
allowing the imine lone pair to abstract a proton from the amino ligand, formally
11
changing it into an amido ligand. The amido ligand and the allyl group can then
reductively eliminate to form a palladium(0) intermediate that is quickly oxidized back to
palladium(II) by protonation from the iminium group, reforming the palladium hydride
intermediate and restarting the catalytic cycle.
Exploring these 3IP complexes further led to the development of [(2diphenylphosphinocyclohexene-1-(2,6-xylyl)imine)Pd(allyl)]OTf (5Pd). Two key ligand
components were varied compared to our first-generation 3IP palladium catalyst,
exchanging the tert-butyl imine group for a 2,6-xylyl moiety and the cyclopentene
backbone ring for the slightly larger cyclohexene ring. It was hypothesized that the
sterically larger xylyl moiety would lead to faster imine decoordination, leading to easier
coordination of amine and thus more productive turnover frequency. This second
generation complex was found to hydroaminate 1,2- and 1,3-dienes with secondary
amines readily.
It produced solely the thermodynamic (linear) product when
hydroaminating the unsymmetrical 1,1-dimethylallene. It did this much more readily than
the first generation 3IP catalyst was capable, hydroaminating 1,1-dimethylallene with
morpholine at room temperature in over 98% conversion, whereas the first generation
catalyst was only capable of a mixture of kinetic (branched) and thermodynamic products
totaling 89% conversion. This was a marked improvement for our catalyst system, which
ultimately led to the synthesis of our third generation 3-iminophosphine palladium
complex.
Our third generation catalyst, [(2-di-tert-butylphosphinocyclopentene-1-(tertbutyl)imine)Pd(allyl)]OTf (4Pd), showed even higher activity for the hydroamination of
allenes at room temperature with regioselectivity unobserved for previous complexes
12
synthesized by our group. This complex hydroaminated 1,1-dimethylallene with a
sterically and electronically diverse set of anilines to form solely the kinetic (branched)
product at room temperature, a catalytic behavior that has yet to be reported by any group
but our own with a palladium centered catalyst system. Electron rich anilines were
converted to hydroamination products in the greatest conversions, whereas halogenated
anilines required mild heating (70 oC) to achieve catalysis. Sterically hindered (orthosubstituted) anilines were not amenable to this reaction. These results further supported
the notion that coordination of the amine/decoordination of the imine is an important step
in the catalytic cycle. Witnessing extremely varied catalytic behavior from what one
would imagine to be the same catalytic system with seemingly inconsequential changes
to the ligand, an array of these 3-iminophosphine complexes that varied systematically in
their ligand structural features was synthesized, characterized, and screened for their
hydroamination catalytic activity. This investigation constitutes the primary investigation
discussed in this thesis.
13
Chapter 2
Investigation of Steric and Electronic
Features of 3-Iminophosphine-Based
Palladium Hydroamination Catalysts
2.1 Introduction
Hydroamination is an advantageous method for the preparation of primary,
secondary, and tertiary amines due to its 100% atom economy1 and the wide range of
regio-, stereo-, and enantioselective metal complexes that are available to catalyze this
transformation.2-10 The hydroamination process is characterized by the addition of an NH bond of ammonia or of a primary or secondary amine across an unsaturated C-C bond
of an alkene, alkyne, or allene. The reaction can take place either intermolecularly at a
high
entropic
demand
using
affordable
and
readily
obtained
substrates
or
intramolecularly with low entropic demand, but requiring less accessible starting
materials. Although hydroamination is slightly exothermic, the direct [2+2] cycloaddition
of an N-H across a C-C unsaturated bond is orbitally forbidden under thermal conditions,
making a catalytic route for this process a virtual necessity.2
14
The hydroamination of allenes allows for the synthesis of allylic amines, which
can be further transformed using a wide variety of reactions including hydroboration,89
hydroformylation,90 alkene metathesis,91 and heterocycle synthesis.2 Allylic amines also
play a major role in the synthesis of pharmaceutical and natural products.92 Early
transition metal complexes have been shown to catalyze allene hydroamination with
addition of the nitrogen group to the central carbon of the allene and subsequent
tautomerization of the resulting enamine to form an imine in the case of primary amine
substrates, whereas late transition metal catalysts are known to hydroaminate allenes with
addition of the nitrogen moiety to one of the terminal carbons, resulting in retention of
the allyl group.2 Intramolecular hydroamination of allenes leads to nitrogen-containing
heterocycles. Although intramolecular hydroamination of allenes has been studied
briefly,59,60,62,63,75-78 with a focus on enantioselective conversion to products, examples of
intermolecular allene hydroamination are more prevalent and include those published by
Bertrand,5,21,57 Widenhoefer,20,79,80 Yamamoto,22,58 Toste,12 and Schafer,18,81 as well as
our own group.3,71 Late transition metal catalyzed intermolecular hydroamination of
substituted allenes yields two possible regioisomers. Addition of the amino moiety to the
substituted carbon terminus of a mono-substituted allene yields a new chiral carbon
center and a vinyl-terminated product, which is typically the kinetic product of these
reactions (Scheme 1-2). Addition of the amine at the less-substituted carbon atom yields
an internal alkene, commonly the thermodynamic product (Scheme 1-2). Regioselective
hydroamination of substituted allenes to form the thermodynamic product is much more
common and has been accomplished previously with aryl amines and a gold catalyst,20,58
secondary alkylamines and gold21,22 or platinum79 catalysts, ammonia and a gold
15
catalyst,57 and hydrazine with a gold catalyst.5 Also notable is Toste’s mechanistic study
involving the hydroamination of a symmetrical allene with methyl carbazate, which
demonstrated activation of the allene as the rate-limiting step and implied that there was
no coordinated amine in the catalytic mechanism.12 Regioselective intermolecular
hydroamination of substituted allenes to form the kinetic (branched) product, though
much rarer, has been accomplished via a gold(I) N-heterocyclic carbene complex,
although substrate scope was limited to the addition of an N-unsubstituted carbamate,4 as
well as with a rhodium(I) Josiphos complex that was able to add a series of anilines
enantioselectively to the more substituted side of the allene.19 The development of new
catalysts capable of producing the branched product in these reactions would be
considerably advantageous in order to expand upon this limited substrate scope and to
allow a much larger variety of allylic amines to be synthesized via hydroamination. Also
of interest is the development of more cost-effective metal catalysts, as gold, platinum,
and rhodium are among the most expensive of the transition metals.
Synthesized previously in our group were 3-iminophosphine (3IP) ligands 2,88 4,3
and 571 (Figure 2-1), as well as their respective [(3IP)Pd(allyl)]OTf complexes 2Pd,88
4Pd,3 and 5Pd.71 2Pd was shown to hydroaminate phenylacetylene and 1,3cyclohexadiene in moderate yields,88 whereas 5Pd was shown to hydroaminate 3-methyl1,2-butadiene (1,1-dimethylallene) and 2,3-dimethyl-1,3-butadiene with secondary
alkylamines regioselectively to form the thermodynamic (linear) products in moderate to
excellent yields.71 The most significant advance in the development of these catalysts
came with the discovery of 4Pd, which was found to regiospecifically hydroaminate 1,1dimethylallene using a variety of aryl amines (anilines) to form solely the kinetic
16
(branched) products in good to excellent yields.3 Because the observed catalysis of these
three different ligand frameworks varied dramatically, it was hypothesized that the
specific steric and electronic features of the ligand were responsible for the greatly
varying catalytic activity. Unfortunately, in the development of our hydroamination
catalysts (2Pd, 4Pd, and 5Pd), the ligand framework was not altered in a systematic
fashion, but rather involved changes to two ligand components with each new catalyst
explored.
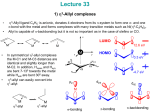
Figure 2-1 3-Iminophosphine (3IP) ligands 1-10.
Thus,
the
catalytic
data
generated
with
our
three
previously
published
[(3IP)Pd(allyl)]OTf complexes was insufficient in determining which alterations to
ligand design were critical in order to optimize catalytic activity. As a means to address
this deficiency, synthesized herein is an array of eight [(3IP)Pd(allyl)]OTf complexes
with systematic variance of the steric and electronic features of the 3IP ligands. Each
palladium complex was screened for catalytic activity in the hydroamination of 1,1 17
dimethylallene with a selection of seven aryl amines spanning a wide range of steric and
electronic parameters. Also reported herein is the synthesis of two additional ligands and
their respective [(3IP)Pd(allyl)]OTf complexes whose designs were based upon the
results of the catalytic screening of the original array of eight complexes.
2.2 Results and Discussion
The effectiveness of 3-iminophosphine (3IP) allylpalladium triflate complexes as
catalysts for the hydroamination of 1,3-dienes, allenes, and alkynes with primary and
secondary amines has been shown previously.71 These results implied that the steric and
electronic properties of the 3IP supporting ligands have a significant effect on the rates
observed in hydroamination catalysis. These ligand properties can be governed via
substitution of three tunable domains: the size of the alicyclic backbone and the
substituent groups of the imine and phosphine moieties. For example, cyclic ketones that
vary in ring size can be reacted with the Vilsmeier-Haack reagent, various amines can be
used for the Schiff base condensation, and phosphination can be completed with different
di-substituted lithium phosphides in the synthesis of these ligands (Scheme 2-1). Our
previous reports detail the synthesis and isolation of ligands 2,88 4,3 and 5,71 as well as
their respective allyl palladium triflate complexes 2Pd,88 4Pd,3 and 5Pd.71 Unfortunately,
the ligands described in our previous reports varied two tunable domains with each
catalyst improvement. The synthesis of complexes with systematic variation of the three
tunable domains would allow for correlation of catalytic activity with each domain
involved in ligand construction.
18
Scheme 2-1 General synthesis of [(3IP)Pd(allyl)]OTf complexes.
(n = 1-4, R’ = tBu or 2,6-dimethylphenyl, R” = tBu or Ph)
(i) 2 eq. DMF, 1.6 eq. POCl 3 , 0 oC, 4 hours then 55 oC, 14 hours; 0 oC, NaHCO 3 ;
(ii) 1.3 eq. H 2 NR’, pentane, 4 Å molecular sieves, 0 oC; (iii) 1.6 eq. LiPR” 2 , Et 2 O, 3
hours, RT. Thus, in the current contribution, we set out to produce a complete series of 3IP ligands
utilizing both backbone ring sizes (cyclopentenyl and cyclohexenyl), imines (tert-butyl
and 2,6-xylyl), and phosphines (tert-butyl and phenyl) to determine the impact of each
unit on catalytic activity (1-8, Figure 2-1). Then, by use of the palladium complexes of
these eight ligands in catalysis, we hoped that an empirical understanding of the
effectiveness of these ligand components would allow for the rational design of improved
hydroamination catalysts.
Compounds 1, 3, and 6 were synthesized using procedures analogous to those for
3-iminophosphines 2,88 4,3 and 5,71 respectively, while ligands 7, 8, 9, and 10 required
19
significant modifications to the experimental methodology (Scheme 2-1). The main
change involved the dropwise addition of a dilute solution of lithium di-tertbutylphosphide in diethyl ether to a rapidly stirred solution of the selected chloroimine in
diethyl ether at ambient temperature. The attempted use of our previous synthetic
procedures consistently resulted in multiple unidentifiable phosphorus containing
products that were inseparable from both the target ligand and the final palladium
complex. Overall, the syntheses of ligands 7-10 were especially sensitive, requiring the
purest of starting materials. Chloroaldehyde of high purity was produced via neat reaction
of the cyclic ketone with the Vilsmeier-Haack reagent (Scheme 2-1), under a nitrogen
atmosphere using degassed reagents. After reacting for 14 h at 55 oC, the resulting
solution was made basic and extracted with pentane, in contrast to diethyl ether as cited
previously.88 This improved methodology, analogous to that of Paquette,93 is vital for
achieving reaction completion and ensuring chloroaldehyde purity, as well as improving
product stability at both low and ambient temperatures. Prior to its use, lithium di-tertbutylphosphide was recrystallized from diethyl ether and rinsed with pentane to minimize
highly reactive impurities that were found to complicate phosphination of the various
chloroimines, leading to multiple unidentifiable phosphorus containing products. Ligand
coordination and ion exchange reactions yielding the final [(3IP)Pd(allyl)]OTf complexes
proceeded analogously to Beck and Schmidt3 (Scheme 2-1) with only slight workup
modifications. Specifically, the intermediate 3IP allylpalladium chloride complex was
rinsed of excess ligand before treatment with silver triflate because we have found that
byproduct silver chloride is readily coordinated by excess 3IP ligand. Furthermore,
toluene was excluded from the anion exchange step, as its presence seems to destabilize
20
the palladium complex, resulting in presumed palladium(0) byproducts that were not
soluble in the readily available solvents.
All of the ligands and palladium complexes described in this chapter are
diamagnetic, and so an analysis of their NMR spectroscopic features proves quite
insightful. As one would anticipate, substantial differences were noted between 1H, 13C,
and
31
P NMR spectra of the free 3-iminophosphine ligands and those of the
[(3IP)Pd(allyl)]OTf complexes. Further differences were evident when comparing
ligands and their respective complexes with di-tert-butylphosphino moieties to those
possessing diphenylphosphino moieties. Downfield shifts in the
31
P NMR signals were
observed upon coordination of the ligand to the palladium, with a further downfield shift
upon
chloride
abstraction
(Table
2.1).
Table 2.1 Comparison between selected 31P and 1H NMR resonances of
[(3IP)Pd(allyl)]OTf complexes and their respective free 3IP ligands (all values in
ppm).
3IPb
Δ31P δ
3IPPda
3IPb
Δ1H δ
3IPPda
31
1
1
Pδ
Pδ
R'N=CH H δ R'N=CH H δ
13.11
-23.20
36.31
7.88
8.64
-0.76
1
16.90
-24.70
41.60
7.94
8.72
-0.78
2
59.48
13.24
46.24
7.84
8.82
-0.98
3
61.80
13.20
48.60
8.06
8.88
-0.82
4
25.20
-12.60
37.80
7.70
9.05
-1.35
5
31.78
-12.70
44.48
7.65
9.09
-1.44
6
71.63
20.05
51.58
7.65
9.32
-1.67
7
75.37
19.98
55.39
7.85
9.37
-1.52
8
80.64
24.81
55.83
8.07
9.37
-1.30
9
55.18
11.36
43.82
7.94
8.27
-0.33
10
a
b
3IPPd refers to [(3IP)Pd(allyl)]OTf complex. 3IP refers to uncoordinated ligand.
31
The absolute change in ppm for the 31P NMR signal between free ligands and the target
[(3IP)Pd(allyl)]OTf complexes (Δ31Pδ) varied greatly depending on the ligand used.
Δ31Pδ was always larger for complexes with larger backbone size while holding
21
phosphine and imine substituents constant. Also, in every case complexes with di-tertbutylphosphino moieties had larger Δ31Pδ values than the corresponding complexes with
diphenylphosphino moieties. Furthermore, all
31
P NMR resonances for complexes and
ligands with di-tert-butylphosphino moieties were located further downfield than those of
the corresponding diphenylphosphino moieties. Keeping imine and phosphine moieties
constant, all complexes with larger backbone sizes displayed signals that were also
located further downfield than those with smaller backbone sizes. These
31
P NMR
spectroscopic trends imply that the phosphorus of the ligand is more tightly bound to the
metal for the larger backbone rings and for the di-tert-butylphosphino complexes. All 1H
NMR resonances for the imine C-H of the palladium complexes rested at or slightly
below 8 ppm, an upfield shift from that of the free ligands, which were typically found
around 9 ppm. Ligand 10 displayed a significantly upfield imine proton resonance
compared to the rest of the free ligands. It also showed the smallest change in 1H NMR
resonance (Δ1Hδ) upon coordination to form complex 10Pd, with a mere -0.33 ppm shift
(the negative number denoting an upfield shift from ligand to triflated complex). The
other species shifted from -0.76 to -1.67 ppm upon formation of the triflate complexes. In
general, the upfield shift in the imine proton resonance can be attributed to reduction in
the carbon-nitrogen double bond character upon coordination of the imine lone pair to
palladium. A smaller than normal upfield shift correlates to a smaller loss of double bond
character consistent with weaker imine nitrogen coordination to palladium. Thus, it
seems that ligand 10 binds less strongly to the metal center through its imine nitrogen
atom than the other ligands in this series. The trends in the
13
C NMR spectra parallel
those of the 1H NMR spectra, as there are significant changes in the chemical shift of the
22
imine carbon between free and coordinated ligands. The terminal carbons of the allyl
ligands (cis and trans to phosphorus) were readily differentiated based upon both the
magnitude of the J P-C coupling constant, and their relative chemical shift values. The
trans carbon was typically located downfield from the cis carbon and displayed a J P-C
coupling constant an order of magnitude larger.
Many of the palladium allyl triflate complexes (1Pd, 2Pd,88 4Pd,3 5Pd,71 8Pd,
and 9Pd) were structurally characterized via X-ray crystallography. In all cases, X-ray
quality single crystals were grown from pentane-layered THF solutions at -25 oC. Each
complex had an outer-sphere triflate anion with palladium’s coordination sphere occupied
by the chelating 3-iminophosphine ligand and a single 3η-allyl group, producing a
distorted square planar geometry. Structures of 1Pd (Figure 2-2) and 5Pd,71 differing
only in the size of the alicyclic backbone, both exhibited disorder in their allyl ligands to
the extent that no discernable comparison could be made between their bite angles. The
series of three complexes with tert-butyl groups on both nitrogen and phosphorus, but
with variation of the backbone size from five to seven carbon atoms, varied less
dramatically in bite angle and seemed more dependent on the position of the allyl group
relative to that of the alicyclic backbone. Viewing the molecule along the P-Pd-N plane,
an allyl group pointing in the direction of the backbone is denoted as cis while one
pointing in the opposite direction is denoted as trans. Complex 4Pd exists as the trans
isomer, 8Pd exists as the cis isomer (Figure 2-3), and 9Pd has both the cis and trans
isomers in its asymmetric unit (Figures 2-4 and 2-5).
23
Figure 2-2. ORTEP (50% thermal ellipsoids) of 1Pd. Triflate anion and hydrogen
atoms have been omitted for clarity. Selected bond lengths (in Å): Pd1-P1 =
2.2680(8), Pd1-N1 = 2.101(2), Pd1-C27 = 2.104(3), Pd1-C29 = 2.236(3), P1-C6 =
1.799(3), C1-C2 = 1.458(4), C2-C6 = 1.346(4), N1-C1 = 1.275(4). Bond angles (in
deg): P1-Pd1-N1 = 95.11(7), C27-Pd1-C29 = 67.5(1), N1-Pd1-C27 = 168.7(1), N1Pd1-C29 = 101.3(1), P1-Pd1-C27 = 96.15(9), P1-Pd1-C29 = 163.59(9), Pd1-P1-C6
= 110.7(1), P1-C6-C2 = 125.6(2), C1-C2-C6 = 130.5(3), N1-C1-C2 = 128.2(3), Pd1N1-C1 = 129.4(2).
24
Figure 2-3. ORTEP (50% thermal ellipsoids) of 8Pd. Triflate anion, disordered tertbutyl carbons, and hydrogen atoms have been omitted for clarity. Selected bond
lengths (in Å): Pd1-P1 = 2.332(2), Pd1-N1 = 2.116(6), Pd1-C12 = 2.208(8), Pd1C14 = 2.146(7), P1-C1 = 1.844(7), C1-C6 = 1.35(1), C6-C7 = 1.49(1), N1-C7 =
1.26(1). Bond angles (in deg): P1-Pd1-N1 = 89.8(2), C12-Pd1-C14 = 66.1(4), N1Pd1-C12 = 100.7(3), N1-Pd1-C14 = 165.3(3), P1-Pd1-C12 = 165.4(2), P1-Pd1-C14
= 104.4(2), Pd1-P1-C1 = 101.7(2), P1-C1-C6 = 122.1(6), C1-C6-C7 = 126.5(7), N1C7-C6 = 129.8(7), Pd1-N1-C7 = 121.6(5).
25
Figure 2-4 ORTEP diagram (50% thermal ellipsoids) of trans-9Pd (top view).
Triflate anion and hydrogen atoms have been omitted for clarity. Selected bond
lengths (in Å): Pd1-P1 = 2.331(1), Pd1-N1 = 2.098(4), Pd1-C21 = 2.162(5), Pd1C23 = 2.257(5), P1-C3 = 1.845(5), C1-C2 = 1.472(6), C2-C3 = 1.361(6), N1-C1 =
1.288(6). Bond angles (in deg): P1-Pd1-N1 = 92.1(1), C21-Pd1-C23 = 66.4(2), N1Pd1-C21 = 166.8(2), N1-Pd1-C23 = 101.5(2), P1-Pd1-C21 = 100.9(1), P1-Pd1-C23
= 162.5(1), Pd1-P1-C3 = 109.1(2), P1-C3-C2 = 122.4(4), C1-C2-C3 = 126.2(4), N1C1-C2 = 131.7(4), Pd1-N1-C1 = 123.2(3). 26
N2
Pd2
P2
P1
Pd1
N1
Figure 2-5 ORTEP diagrams (50% thermal ellipsoids) of cis (top) and trans
(bottom) isomers of 9Pd (side views). Triflate anion, hydrogen atoms, and tert-butyl
groups have been omitted for clarity.
27
Those with the trans relationship had more strained bite angles, i.e. 4Pd at
92.52(6)o and trans-9Pd at 92.1(1)o, while the cis complexes 8Pd and cis-9Pd had bite
angles of 89.8(2)o and 90.1(1)o, respectively. Again, the cyclopentenyl complex (4Pd)
deviated the most from the ideal 90o bite angle. Complex 2Pd, which is a hybrid of the
two series, has a bite angle of 92.9(5)o, very close to that of 4Pd, most likely due to its
similar trans configuration. This may also indicate that the imine’s substituent group is
more significant in determining the angle than those of the phosphine. All Pd-N distances
varied little, whereas Pd-P distances were generally shorter for diphenylphosphino
moieties (~2.27 Å) than for di-tert-butylphosphino moieties (~2.33 Å), which likely
reflects the larger steric bulk of the tert-butyl groups. Similar disubstituted bidentate
phosphine palladium complexes show similar Pd-P bond distance patterns.94,95
In our most recent report, compound 4Pd was shown to readily catalyze the
hydroamination of 1,1-dimethylallene with primary aryl amines (anilines) to form
exclusively the kinetic (terminal alkene) products at room temperature (Product A; Table
2).3 Sterically hindered anilines prevented hydroamination in all cases, while halogenated
anilines required heating to a temperature of 70 oC in order to achieve appreciable
conversion to product. In an effort to more fully understand this reactivity, to improve
upon the catalytic performance of 4Pd, and to test our previously reported complexes
2Pd88 and 5Pd71 in allene hydroamination, the complete array of ligands 1-8 and their
respective complexes 1Pd-8Pd was synthesized. All eight palladium complexes were
then tested in the hydroamination of 1,1-dimethylallene with a selection of seven anilines
displaying diverse steric and electronic properties (Table 2.2).
28
The results generated by screening these eight palladium complexes were
generally disappointing, as most of these complexes showed no catalytic turnover for the
hydroamination of 1,1-dimethylallene with anilines. From this array, only our previously
reported 4Pd was found to catalyze this hydroamination effectively. Complex 2Pd also
catalyzed the reaction, albeit with extremely poor conversions compared to that of 4Pd.
Complexes 2Pd and 4Pd are very similar, differing only in the substituent groups of their
phosphine moieties. The phosphine tert-butyl groups of 4Pd seem to play a vital role in
its catalytic activity, as substituting them with phenyl groups in 2Pd virtually eliminated
catalytic
activity.
Table 2.2 Conversion to product A for the [(3IP)Pd(allyl)]OTf catalyzed
hydroamination of 1,1-dimethylallene with aryl amines (complexes 1Pd-10Pd).
Conversion was monitored via 1H NMR spectroscopy.
Aniline
1Pd
2Pd
3Pd
4Pd
5Pd
6Pd
7Pd
8Pd
9Pd
10Pd
--
--
--
60%
--
--
--
--
--
91%
--
--
--
--
--
--
--
6%
--
92%
--
--
--
--
>95%
--
--
--
--
>95%
--
--
--
--
90%
--
--
6%
--
72%
2-Me
-----Aniline
3-Me
-13% ->95% -Aniline
4-Me
-16% ->95% -Aniline
4-tBu
-17% -88%
-Aniline
4-F
-9%
-65%* -Aniline
3-OMe -12% -62%
-Aniline
* indicates reaction at 70oC. “--” indicates <5%.
29
Changing the tert-butyl group of the imine moiety into the quite sterically-hindered 2,6dimethylphenyl group resulted in complete loss of catalytic activity in all four complexes
containing that substituent (1Pd, 3Pd, 5Pd, 7Pd). Furthermore, increasing the backbone
ring size to a cyclohexene ring also destroyed catalytic activity across the series of
complexes (see 5Pd-8Pd). Overall, as is so often noted in catalyst design, a very specific
set of features (found in 4Pd) were crucial to achieve catalysis, with minor changes
having a devastating impact on catalytic activity.
Despite the fact that no improved catalysts were discovered upon screening the
array of 1Pd-8Pd, we were convinced that this method would yield an improved catalyst
and so further investigation into ligand design was undertaken. The results observed for
complexes 1Pd-8Pd necessitated that further ligand designs incorporate tert-butyl groups
on both the phosphine and imine moieties of the 3IP ligands. Believing that the
conformational stability of the cyclohexene ring was the downfall of 8Pd when compared
to the less stable cyclopentene ring of 4Pd, ligand 9 and complex 9Pd (each bearing a
cycloheptenyl ring) were synthesized. Unfortunately, the conformational effects of the
cycloheptene ring of 9Pd did not grant any greater catalytic performance than that of the
cyclohexene ring. Thus, we explored the opposite approach, utilizing the smaller and
highly strained cyclobutenyl ring, leading to the synthesis of 10 and 10Pd. The smaller
alicyclic ring size of 10Pd significantly enhanced catalytic activity, producing the same
regiospecific product as 4Pd, but at higher conversions for virtually every substrate and
at lower temperature for those that had required heating with 4Pd. Most notably, 10Pd
catalyzed the reaction of 4-fluoroaniline with 1,1-dimethylallene at room temperature to
give 90% conversion, as compared to the 65% conversion at 70 oC when using 4Pd.
30
Also, the reaction of unsubstituted aniline was also significantly improved, converting
91% of substrate to product, a 31% increase over that previously reported.3 Due to the
superior performance of 10Pd compared to all of the other 3IP complexes investigated
thus far, it is clear that the size of the alicyclic ring is a critical component in the design
of [(3IP)Pd(allyl)]OTf catalysts, with the cyclobutenyl backbone proving to be preferred
over larger ring sizes.
2.3 Conclusions
A collection of ten [(3IP)Pd(allyl)]OTf complexes was investigated in the
catalytic hydroamination of 1,1-dimethylallene with electronically and sterically diverse
aryl amines. There is a strong correlation between the three tunable ligand structural
domains and the catalytic activity of these complexes. The ligand composed of di-tertbutyl phosphine, cyclobutenyl backbone, and tert-butyl imine domains led to the most
active palladium catalyst when compared to the other ligands in this collection. Better
electron donating substituents on the phosphine and imine moieties grant greater catalytic
ability to these complexes while larger alicyclic backbone ring sizes almost completely
eliminate catalytic activity. Excellent regiospecific hydroamination of 1,1-dimethylallene
is now attainable for all but the most sterically hindered anilines. Further refinement of
the 3IP ligand set, including investigation into the effects of phosphine and imine
moieties of even greater electron donating character is ongoing. Additionally, use of these
palladium complexes in other catalytic transformations, as well as one-pot multistep
syntheses, continues to be under investigation in our laboratory.
31
2.4 Experimental Section
2.4.1 General Methods and Instrumentation
Alicyclic α,β-unsaturated β-chloroaldehydes and β-chloroimines were synthesized
under ambient atmospheric conditions. All other manipulations were performed under an
inert N 2 atmosphere using standard Schlenk and drybox techniques. Solvents were
predried prior to use; methylene chloride was passed through two columns of 4 Å
molecular sieves and degassed with nitrogen. Pentane, diethyl ether, and toluene were
passed through columns of activated alumina and 4 Å molecular sieves and degassed
with nitrogen. Tetrahydrofuran was distilled from sodium metal and degassed with
nitrogen. n-Butyllithium (1.6 M in hexanes), (allyl)palladium(II) chloride dimer,
diphenylchlorophosphine, ditert-butylchlorophosphine, lithium aluminum hydride, and
silver triflate were purchased from Strem and used without further purification.
Phosphorus oxychloride, tert-butylamine, 2,6-dimethyl aniline, and cyclopentanone were
purchased from Acros and used without further purification. Cyclohexanone, 1,1dimethylallene and cycloheptanone were purchased from Alfa Aesar and used without
further purification. Cyclobutanone was purchased from Sigma Aldrich and used without
further purification. Dimethylformamide was purchased from BDH and stored over 4 Å
molecular sieves. Anilines were purchased from Sigma-Aldrich or another commercial
source and dried over calcium hydride, either neat (liquid anilines) or as solutions in
methylene chloride (solid anilines). Liquid anilines were freeze-pump-thawed three times
and vacuum distilled. Solutions of solid anilines in methylene chloride were freezepump-thawed three times, filtered, and the methylene chloride removed via reduced
pressure. CDCl 3 was purchased from Cambridge Isotope Laboratories, vacuum
32
transferred from CaH 2 , and stored over 4 Å molecular sieves. Benzene-d 6 was also
purchased from Cambridge Isotope Laboratories, vacuum transferred from sodium metal,
and stored over 4 Å molecular sieves. Silica gel (Porosity: 60 Å, Particle size: 40-63 μm)
was purchased from Sorbent Technologies and used as received. 1H and
13
C NMR data
were obtained on a 600 MHz Inova or 400 MHz VXRS NMR spectrometer at ambient
temperature at 599.9 MHz for 1H NMR and 150.8 MHz for 13C NMR and 399.95 MHz
for 1H NMR and 100.56 MHz for
13
C NMR, respectively. All
31
P NMR spectra were
collected on a 400 MHz VXRS NMR spectrometer at ambient temperature at 161.90
MHz. All spectra were taken using C 6 D 6 or CDCl 3 as the NMR solvent. 1H NMR shifts
are given relative to the residual solvent resonances at 7.16 and 7.26 ppm, respectively,
and 13C NMR shifts are given relative to the residual solvent peak of CDCl 3 (77.36 ppm).
31
P NMR spectra were externally referenced to 0.00 ppm with 5% H 3 PO 4 in D 2 O. IR
samples were prepared as Nujol mulls and taken between KBr plates on a Perkin-Elmer
XTL FTIR spectrophotometer. Melting points were observed on a capillary melting point
(Uni-Melt) apparatus in sealed capillary tubes and are uncorrected. X-ray structure
determinations were performed at the Ohio Crystallographic Consortium, housed at The
University of Toledo. Elemental analyses were determined by Atlantic Microlab, Inc.,
Norcross, GA or Galbraith Laboratories, Inc., Knoxville, TN. High resolution mass
spectrometry using electrospray ionization was performed at the University of Illinois
Mass Spectrometry Laboratory, Urbana, IL.
33
2.4.2 General Procedure for the Catalytic Hydroamination Screening of Compounds
1Pd-10Pd
All manipulations were performed under an N 2 atmosphere. 3-Methyl-1,2butadiene (68 mg, 1 mmol) was added to a mixture of amine (0.5 mmol),
[(3IP)Pd(allyl)]OTf complex (5 mol%), and deuterated benzene (0.8 ml). Conversion to
products was monitored via 1H NMR spectroscopy. Hydroamination products formed
were reported previously.3
2.4.3 Synthesis and Characterization of Reaction Products
The following compounds were synthesized as previously reported: LiPPh 2 ,88 LiPtBu 2 ,3
2-chlorocyclopentenecarboxaldehyde,88
2-chlorocyclohexenecarboxaldehyde,71
2-
chlorocyclohexene-1-(2,6-xylyl)imine,71 2-chlorocyclopentene-1-(tert-butyl)imine,88 2diphenylphosphinocyclopentene-1-(tert-butyl)imine
(2),88
2-di-tert-
butylphosphinocyclopentene-1-(tert-butyl)imine (4),3 2-diphenylphosphinocyclohexene1-(2,6-xylyl)imine
(5),71
butyl)imine)Pd(allyl)]OTf
(2Pd),88
butyl)imine)Pd(allyl)]OTf
(4Pd),3
[(2-diphenylphosphinocyclopentene-1-(tert[(2-di-tert-butylphosphinocyclopentene-1-(tertand
[(2-diphenylphosphinocyclohexene-1-(2,6-
xylyl)imine)Pd(allyl)]OTf (5Pd).71
Alicyclic α,β-Unsaturated β-Chloroaldehydes
The following was performed with degassed reagents and solvents, and an atmosphere of
nitrogen was maintained over the mixture as the reaction proceeded. Dimethylformamide
(9.00 g, 123 mmol) was cooled to 0 oC. POCl 3 (15.00 g, 97 mmol) was then added
34
dropwise with rapid stirring. Formation of the Vilsmeier-Haack reagent was allowed to
proceed for 4 h, slowly warming to room temperature. The reagent was again cooled to 0
o
C before dropwise addition of the respective cyclic ketone (61 mmol). After slowly
warming to room temperature, the mixture was heated at 55 oC for 14 h. The crude
reaction mixture was quenched over water at 0 oC to produce the chloroaldehyde. The
mixture was made basic with an excess of sodium bicarbonate before extracting into
pentane (4 × 100 ml). The organic layer was rinsed with water and brine and dried over
magnesium sulfate for 20 min. The organic layer had ethyl acetate added up to 2% and
was passed through a plug of silica. Solvent was evaporated to yield the final product as a
lightly colored liquid.
Alicyclic α,β-Unsaturated β-Chloroimines
tert-Butyl imines
To pentane (20 ml) was added activated 4 Å molecular sieves. This was cooled to 0 oC
before adding freshly prepared chloroaldehyde and 1.3 equivalents of tert-butylamine.
The reaction was allowed to proceed for 14 h, slowly warming to ambient temperature.
Magnesium sulfate was added to the reaction mixture and allowed to stir for 20 min
before filtering over celite. Diethyl ether and excess tert-butylamine were evaporated,
yielding the final product.
2,6-Dimethyl phenyl imines
To pentane (20 ml) was added activated 4 Å molecular sieves. This was cooled to 0 oC
before adding freshly prepared chloroaldehyde and 1.3 equivalents of 2,6-
35
dimethylaniline. The reaction was stirred for 96 h, slowly warming to ambient
temperature. Magnesium sulfate was added to the reaction mixture and allowed to stir for
20 min before filtering over celite. Diethyl ether was evaporated and the product
redissolved in pentane. Ethyl acetate was added up to 2% and the product solution was
passed through a plug of silica to remove excess aniline. Solvent was evaporated,
yielding the final product.
3-Iminophosphine Ligands
Unless otherwise noted, all 3-iminophosphine ligands were prepared via the following
methodology. To a Schlenk tube with attached addition funnel was added degassed
chloroimine (1 equiv) and diethyl ether (15 ml). Lithium di-tert-butylphosphide (1.6
equiv) was dissolved in diethyl ether (30 ml) in a separate flask. The lithium phosphide
was transferred to the addition funnel and then added dropwise to the rapidly stirring
chloroimine solution at ambient temperature over a time period of 1 h. The reaction was
allowed to stir for an additional 2 h before concentration in vacuo to an approximate
volume of 10 ml. At this time, pentane (20 ml) was added to help precipitate excess
lithium phosphide. The supernatant was separated via cannula filtration, solvent removed
in vacuo, and the resulting oil triturated with pentane (2 × 10 ml). The oil was then
extracted into pentane (2 × 20 ml) and passed through a celite padded frit. Solvent was
again removed in vacuo to yield the final product.
36
(3-Iminophosphine)allylpalladium Triflate Complexes
Solutions of both ligand (1.05 equiv) and allylpalladium chloride dimer (0.5 equiv) in
dichloromethane (10 ml each) were combined at ambient temperature and allowed to stir
for 14 h. Solvent was removed in vacuo, and the crude solid was triturated and rinsed
with pentane (10 ml each). The solid was redissolved in methylene chloride (10 ml) and
added to a slurry of silver triflate (0.65 equiv) in methylene chloride (10 ml) and stirred
in the dark for 14 h. The crude reaction mixture was then passed through a thick pad of
celite, and solvent removed in vacuo. The resulting solid was triturated with pentane (10
ml), dissolved in a minimal amount of tetrahydrofuran, layered with pentane, and cooled
to -20 oC to promote crystal growth.
2-Chlorocycloheptenecarboxaldehyde:
light yellow liquid (6.128 g, 63.18%); 1H NMR (CDCl 3 ): δ 10.11 (s, 1H), 2.83-2.80 (m,
2H), 2.49-2.47 (m, 2H), 1.80-1.76 (m, 2H), 1.71-1.65 (m, 2H), 1.49-1.43 (m, 2H);
13
C{1H- NMR (CDCl 3 ): δ 191.1, 156.6, 138.7, 41.9, 31.8, 25.7, 25.1, 24.9; IR: 3331 (w),
2926 (s), 2854 (s), 2740 (w), 2698 (w), 1737 (s), 1675 (s), 1638 (s), 1607 (s), 1446 (s),
1389 (m), 1373 (m), 1337 (m), 1239 (s), 1218 (s), 1140 (s), 1083 (m), 1062 (s), 1047 (m),
1016 (m), 974 (s), 959 (s), 907 (m), 886 (m), 829 (m), 808 (m), 761 (s), 689 (s) cm-1.
2-Chlorocyclobutenecarboxaldehyde:
light yellow liquid (1.39 g, 19.3%); 1H NMR (CDCl 3 ): δ 9.69 (s, 1H), 2.85 (t, 3J H-H = 3.6
Hz, 2H), 2.65 (t, 3J H-H = 3.6 Hz, 2H);
13
C{1H} NMR (CDCl 3 ): δ 183.8, 143.3, 141.5,
35.4, 25.1; IR: 3326 (m), 2978 (s), 2945 (s), 2815 (m), 2717 (w), 1782 (m), 1722 (s),
37
1673 (s), 1607 (s), 1439 (m), 1417 (m), 1373 (s), 1286 (s), 1210 (s), 1112 (s), 965 (s),
878 (m), 791 (w), 769 (m), 731 (s) cm-1.
2-Chlorocyclopentene-1-(2,6-xylyl)imine:
orange-red liquid (3.563 g, 95.77%); 1H NMR (CDCl 3 ): δ 8.23 (s, 1H), 7.05 (d, 3J H-H =
7.8 Hz, 2H), 6.94 (t, 3J H-H = 7.8 Hz, 1H), 2.86 (td, 3J H-H = 7.8 Hz, 2J H-H = 2.1 Hz, 1H),
2.85 (td, 3J H-H = 7.8 Hz, 2J H-H = 2.4 Hz, 1H), 2.82 (td, 3J H-H = 7.8 Hz, 2J H-H = 2.1 Hz,
1H), 2.81 (td, 3J H-H = 7.8 Hz, 2J H-H = 2.4 Hz, 1H), 2.12 (s, 6H), 1.96 (pent, 3J H-H = 7.8
Hz, 2H); 13C{1H} NMR (CDCl 3 ): δ 157.6, 151.4, 141.7, 135.8, 128.2, 127.9, 124.0, 39.9,
30.5, 20.9, 18.6; IR: 3062 (w), 3020 (w), 2957 (s), 2915 (s), 2852 (m), 2727 (w), 2360
(w), 2035 (w), 1918 (w), 1839 (w), 1724 (w), 1656 (m), 1625 (s), 1609 (s), 1593 (s),
1467 (s), 1441 (m), 1378 (m), 1347 (m), 1274 (m), 1247 (m), 1190 (s), 1158 (w), 1090
(m), 1033 (w), 985 (w), 938 (m), 912 (w), 844 (m), 760 (s), 723 (m), 671 (w) cm-1.
2-Chlorocyclohexene-1-(tert-butyl)imine:
yellow-orange liquid (1.626 g, 81.30%); 1H NMR (CDCl 3 ): δ 8.46 (s, 1H), 2.50-2.46 (m,
2H), 2.42-2.38 (m, 2H), 1.77-1.71 (m, 2H), 1.68-1.62 (m, 2H), 1.21 (s, 9H); 13C{1H}
NMR (CDCl 3 ): δ 154.2, 138.7, 131.7, 57.4, 35.2, 29.8, 26.0, 23.7, 21.8; IR: 2964 (s),
2932 (s), 2859 (s), 2356 (w), 2324 (w), 1717 (w), 1691 (w), 1623 (m), 1560 (w), 1455
(w), 1434 (w), 1366 (m), 1266 (w), 1240 (w), 1209 (m), 1172 (w), 1135 (w), 1115 (w),
1099 (w), 1072 (w), 1025 (w), 989 (m), 957 (w), 905 (w), 868 (w), 826 (w), 779 (w), 695
(w) cm-1.
38
2-Chlorocycloheptene-1-(tert-butyl)imine:
light yellow liquid (3.640 g, 50.97%); 1H NMR (CDCl 3 ): δ 8.37 (s, 1H), 2.74-2.71 (m,
2H), 2.67-2.64 (m, 2H), 1.77-1.75 (m, 2H), 1.65-1.62 (m, 2H), 1.50-1.47 (m, 2H), 1.21
(s, 9H); 13C{1H} NMR (CDCl 3 ): δ 155.0, 142.8, 137.4, 57.7, 40.7, 32.0, 30.3, 26.9, 26.1,
25.5; IR: 3217 (w), 2968 (s), 2926 (s), 2698 (w), 1737 (w), 1680 (m), 1618 (s), 1446 (s),
1368 (s), 1332 (m), 1280 (m), 1259 (s), 1213 (s), 1140 (m), 1099 (m), 1083 (m), 1062
(m), 1016 (w), 974 (s), 959 (s), 922 (m), 901 (s), 876 (w), 829 (m), 808 (w), 761 (s), 684
(m), 626 (m) cm-1.
2-Chlorocyclobutene-1-(tert-butyl)imine:
yellow liquid (0.833 g, 40.7%); 1H NMR (CDCl 3 ): δ 7.94 (s, 1H), 2.74-2.73 (m, 2H),
2.66-2.64 (m, 2H), 1.20 (s, 9H);
13
C{1H} NMR (CDCl 3 ): δ 147.4, 141.2, 130.3, 57.9,
34.7, 29.9, 26.0; IR: 3196 (w), 2967 (s), 2935 (s), 2869 (m), 1787 (w), 1673 (m), 1646
(s), 1607 (m), 1515 (w), 1466 (m), 1422 (w), 1390 (w), 1362 (s), 1335 (m), 1297 (m),
1259 (s), 1205 (s), 1112 (s), 1096 (m), 1020 (m), 976 (s), 954 (m), 894 (m), 878 (m), 802
(m), 785 (m), 731 (m), 573 (s) cm-1.
2-Diphenylphosphinocyclopentene-1-(2,6-xylyl)imine (1):
Prepared in a manner analogous to Shaffer et al.88
yellow-orange solid (0.618 g, 61.8%); mp 99-100°C; 1H NMR (CDCl 3 ): δ 8.64 (d, 4J P-H
= 3.6 Hz, 1H), 7.38-7.34 (m, 10H), 7.00 (d, 3J H-H = 7.8 Hz, 2H), 6.90 (t, 3J H-H = 7.8 Hz,
1H), 3.03-3.00 (m, 2H), 2.46-2.44 (m, 2H), 2.04 (s, 6H), 1.96 (pseudo pent, 3J H-H = 7.8
Hz, 3J H-H = 7.2 Hz, 2H); 13C{1H} NMR (CDCl 3 ): δ 159.8 (d, 3J P-C = 22.0 Hz), 153.1 (d,
39
2
J P-C = 21.4 Hz), 151.7, 150.2 (d, 1J P-C = 22.6 Hz), 136.5 (d, 1J P-C = 8.7 Hz), 133.4 (d,
2
J P-C = 19.4 Hz), 129.0, 128.8 (d, 3J P-C = 6.9 Hz), 128.2, 127.2, 123.8, 38.1 (d, 3J P-C = 4.4
Hz), 34.0 (d, 2J P-C = 5.4 Hz), 22.7 (d, 3J P-C = 2.0 Hz), 18.5; 31P{1H} NMR (CDCl 3 ): δ 23.20; IR: 2961 (s), 2919 (s), 2857 (s), 2720 (w), 1612 (m), 1586 (w), 1455 (s), 1376 (m),
1298 (w), 1240 (w), 1193 (m), 1156 (w), 1088 (w), 1025 (w), 999 (w), 968 (w), 915 (w),
842 (w), 764 (m), 738 (m), 722 (m), 696 (m) cm-1. HRMS (m/z): [M+H]+ calcd for
C 26 H 27 NP 384.1882, HRMS found: 384.1874.
2-Di-tert-butylphosphinocyclopentene-1-(2,6-xylyl)imine (3):
Prepared in a manner analogous to that of Beck et al.3
deep red oil (0.910 g, 75.8%); 1H NMR (CDCl 3 ): δ 8.82 (d, 4J P-H = 5.4 Hz, 1H), 7.00 (d,
3
J H-H = 7.8 Hz, 2H), 6.89 (t, 3J H-H = 7.8 Hz, 1H), 2.98-2.95 (m, 2H), 2.88-2.85 (m, 2H),
2.07 (s, 6H), 1.99-1.96 (m, 2H), 1.19 (d, 3J P-H = 11.4 Hz, 18H); 13C{1H} NMR (CDCl 3 ):
δ 161.5 (d, 3J P-C = 29.9 Hz), 156.2 (d, 2J P-C = 20.7 Hz), 152.1, 151.9 (d, 1J P-C = 36.5 Hz),
128.1, 127.1, 123.6, 40.1 (d, 2J P-C = 6.5 Hz), 32.9 (d, 3J P-C = 5.7 Hz), 32.6 (d, 1J P-C = 19.9
Hz), 31.1 (d, 2J P-C = 14.3 Hz), 23.9, 18.7; 31P{1H} NMR (CDCl 3 ): δ 13.24; IR: 2945 (s),
2853 (s), 1608 (s), 1466 (s), 1360 (m), 1324 (m), 1254 (m), 1190 (m), 1169 (m), 1084
(m), 1062 (m), 1013 (m), 843 (w), 801 (m), 758 (s), 716 (w) cm-1. HRMS (m/z): [M+H]+
calcd for C 22 H 35 NP 344.2507, HRMS found: 344.2509.
40
2-Diphenylphosphinocyclohexene-1-(tert-butyl)imine (6):
Prepared in a manner analogous to that of Kuchenbeiser et al.71
off-white liquid (0.824 g, 82.4%); 1H NMR (CDCl 3 ): δ 9.09 (d, 4J P-H = 3.6 Hz, 1H), 7.387.32 (m, 10H), 2.56-2.53 (m, 2H), 1.90-1.88 (m, 2H), 1.66-1.63 (m, 2H), 1.58-1.56 (m,
2H), 1.13 (s, 9H); 13C{1H} NMR (CDCl 3 ): δ 156.4 (d, 3J P-C = 40.2 Hz), 147.6 (d, 2J P-C =
17.7 Hz), 140.7 (d, 1J P-C = 20.4 Hz), 136.7 (d, 1J P-C = 10.8 Hz), 133.6 (d, 2J P-C = 18.9
Hz), 128.6 (d, 3J P-C = 6.3 Hz), 128.6, 57.7, 30.2 (d, 3J P-C = 3.6 Hz), 30.1, 27.0 (d, 2J P-C =
5.8 Hz), 23.7, 22.3; 31P{1H} NMR (CDCl 3 ): δ -12.70; IR: 3126 (w), 3054 (w), 2962 (s),
2921 (s), 2849 (m), 2664 (w), 2274 (w), 1952 (w), 1885 (w), 1814 (w), 1757 (w), 1618
(s), 1582 (w), 1475 (w), 1448 (w), 1428 (s), 1362 (m), 1326 (w), 1305 (w), 1259 (m),
1202 (s), 1090 (m), 1064 (m), 1023 (m), 961 (w), 900 (w), 843 (w), 802 (m), 740 (s), 694
(s) cm-1. HRMS (m/z): [M+H]+ calcd for C 23 H 29 NP 350.2038, found 350.2043.
2-Di-tert-butylphosphinocyclohexene-1-(2,6-xylyl)imine (7):
bright yellow solid (1.100 g, 91.67%); mp 90-91 oC; 1H NMR (CDCl 3 ): δ 9.32 (d, 4J P-H =
9.6 Hz, 1H), 7.01 (d, 3J H-H = 7.2 Hz, 2H), 6.89 (t, 3J H-H = 7.2 Hz, 1H), 2.72-2.70 (m, 2H),
2.66 (br s, 2H), 2.08 (s, 6H), 1.79-1.77 (m, 2H), 1.72-1.69 (m, 2H), 1.21 (d, 3J P-H = 11.4
Hz, 18H); 13C{1H} NMR (CDCl 3 ): δ 164.5 (d, 3J P-C = 48.9 Hz), 151.9, 149.0 (d, 2J P-C =
18.6 Hz), 148.4 (d, 1J P-C = 36.8 Hz), 127.7, 126.9, 123.0, 32.6 (d, 1J P-C = 24.1 Hz), 32.1
(d, 3J P-C = 5.0 Hz), 31.1 (d, 2J P-C = 15.1 Hz), 26.6 (d, 2J P-C = 6.5 Hz), 23.1, 22.2, 18.4;
31
P{1H} NMR (CDCl 3 ): δ 20.05; IR: 2926 (s), 2854 (s), 1612 (w), 1592 (w), 1462 (s),
1379 (m), 1259 (w), 1197 (w), 1171 (w), 1088 (w), 1016 (w), 844 (w), 803 (w), 761 (w),
720 (w) cm-1. HRMS (m/z): [M+H]+ calcd for C 23 H 37 NP 358.2664, found 358.2656.
41
2-Di-tert-butylphosphinocyclohexene-1-(tert-butyl)imine (8):
light brown liquid (0.715 g, 71.5%); 1H NMR (CDCl 3 ): δ 9.37 (d, 4J P-H = 10.8 Hz, 1H),
2.58-2.56 (m, 2H), 2.50-2.48 (m, 2H), 1.68-1.59 (m, 4H), 1.20 (d, 3J P-H = 7.2 Hz, 18H),
1.19 (s, 9H); 13C{1H} NMR (CDCl 3 ): δ 159.2 (d, 3J P-C = 47.1 Hz), 149.5 (d, 2J P-C = 18.1
Hz), 144.1 (d, 1J P-C = 35.4 Hz), 57.4 (d, 5J P-C = 1.4 Hz), 32.2 (d, 3J P-C = 5.0 Hz), 31.4 (d,
2
J P-C = 14.9 Hz), 31.0 (d, 1J P-C = 15.2 Hz), 30.4, 27.5 (d, 2J P-C = 5.0 Hz), 23.5, 22.6;
31
P{1H} NMR (CDCl 3 ): δ 19.98; IR: 2926 (s), 2854 (s), 1618 (m), 1462 (s), 1363 (s),
1259 (m), 1202 (m), 1171 (m), 1088 (m), 1016 (m), 964 (w), 901 (w), 803 (m), 720 (w)
cm-1. HRMS (m/z): [M+H]+ calcd for C 19 H 37 NP 310.2664, found 310.2659.
2-Di-tert-butylphosphinocycloheptene-1-(tert-butyl)imine (9):
yellow oil (0.540 g, 61.6%); 1H NMR (CDCl 3 ): δ 9.37 (d, 4J P-H = 10.8 Hz, 1H), 2.77-2.71
(m, 4H), 1.78-1.75 (m, 2H), 1.62-1.59 (m, 2H), 1.45-1.43 (m, 2H), 1.20 (s, 9H), 1.18 (d,
3
J P-H = 12.0 Hz, 18H);
13
C{1H} NMR (CDCl 3 ): δ 159.4 (d, 3J P-C = 48.6 Hz), 157.3 (d,
2
J P-C = 18.3 Hz), 149.6 (d, 1J P-C = 36.8 Hz), 57.4 (d, 5J P-C = 1.4 Hz), 35.5 (d, 3J P-C = 5.6
Hz), 33.4 (d, 1J P-C = 24.7 Hz), 32.9, 31.4 (d, 2J P-C = 15.2 Hz), 30.5, 29.4 (d, 2J P-C = 6.8
Hz), 28.1, 26.3; 31P{1H} NMR (CDCl 3 ): δ 24.81; IR: 2916 (s), 2854 (s), 1612 (m), 1462
(s), 1363 (s), 1259 (m), 1213 (m), 1171 (m), 1088 (w), 1016 (m), 959 (w), 876 (w), 808
(m) cm-1. HRMS (m/z): [M+H]+ calcd for C 20 H 39 NP 324.2820, found 324.2817.
2-Di-tert-butylphosphinocyclobutene-1-(tert-butyl)imine (10):
brown oil (0.365 g, 68.9%); 1H NMR (CDCl 3 ): δ 8.27 (d, 4J P-H = 2.0 Hz, 1H), 2.87-2.86
(m, 4H), 1.20 (s, 9H), 1.19 (d, 3J P-H = 11.6 Hz, 18H); 13C{1H} NMR (CDCl 3 ): δ 159.1 (d,
42
3
J P-C = 21.6 Hz), 151.4 (d, 1J P-C = 39.8 Hz), 151.0 (d, 2J P-C = 5.3 Hz), 57.7, 33.4 (d, 1J P-C
= 4.7 Hz), 33.0 (d, 2J P-C = 16.5 Hz), 30.7 (d, 2J P-C = 13.3 Hz), 30.1, 29.4 (d, 3J P-C = 11.4
Hz); 31P{1H} NMR (CDCl 3 ): δ 11.36; IR: 2956 (s), 2869 (m), 1628 (w), 1571 (w), 1529
(w), 1472 (m), 1389 (m), 1363 (m), 1259 (s), 1213 (m), 1192 (m), 1176 (m), 1093 (s),
1021 (s), 865 (m), 803 (s), 663 (m) cm-1. HRMS (m/z): [M+H]+ calcd for C 17 H 33 NP
282.2351. found 282.2351.
[(2-Diphenylphosphinocyclopentene-1-(2,6-xylyl)imine)Pd(allyl)]OTf (1Pd):
yellow solid (0.154 g, 92.1%); mp 191 oC dec; 1H NMR (CDCl 3 ): δ 7.88 (d, 4J P-H = 2.4
Hz, 1H), 7.58-7.55 (m, 8H), 7.51-7.48 (m, 2H), 7.11-7.05 (m, 3H), 5.81-5.77 (m, 1H),
3.66-3.62 (m, 1H), 3.55-3.50 (m, 1H), 3.49 (d, 3J H-H = 2.4 Hz, 1H), 3.11-3.04 (m, 2H),
2.70 (d, 3J H-H = 12.0 Hz, 1H), 2.65-2.57 (m, 2H), 2.21 (s, 3H), 2.14-2.08 (m, 2H), 2.10 (s,
3H);
13
C{1H} NMR (CDCl 3 ): δ 164.4 (d, 3J P-C = 7.2 Hz), 155.8, 153.0 (d, 2J P-C = 17.2
Hz), 137.8 (d, 1J P-C = 32.6 Hz), 133.2 (d, 4J P-C = 13.6 Hz), 132.8 (d, 4J P-C = 13.1 Hz),
132.4 (d, 2J P-C = 24.3 Hz), 130.2 (d, 3J P-C = 10.7 Hz), 130.17 (d, 3J P-C = 10.9 Hz), 129.2,
129.0, 128.6 (d, 1J P-C = 50.1 Hz), 128.2 (d, 1J P-C = 49.8 Hz), 127.3, 123.9 (d, 2J P-C = 5.9
Hz), 122.4, 120.2, 87.1 (d, 2J P-C = 28.4 Hz), 55.0 (d, 2J P-C = 3.5 Hz), 39.0 (d, 2J P-C = 11.3
Hz), 36.5, 22.4 (d, 3J P-C = 5.4 Hz), 18.9, 18.8; 31P{1H} NMR (CDCl 3 ): δ 13.11; IR: 2926
(s), 2854 (s), 2719 (w), 2677 (w), 1462 (s), 1379 (s), 1301 (w), 1259 (w), 1218 (w), 1145
(w), 1093 (w), 1031 (w), 969 (w), 798 (w), 720 (w), 637 (w) cm-1. Anal. Calcd for
C 30 H 31 F 3 NO 3 PPdS: C, 52.99; H, 4.60; N, 2.06. Found: C, 52.99; H, 4.57; N, 2.17.
43
[(2-Di-tert-butylphosphinocyclopentene-1-(2,6-xylyl)imine)Pd(allyl)]OTf (3Pd):
dark brown solid (0.736 g, 61.5%); mp 131 oC dec; 1H NMR (CDCl 3 ): δ 7.84 (d, 4J P-H =
1.2 Hz, 1H), 7.14-7.07 (m, 3H), 5.69-5.64 (m, 1H), 4.16 (d, 3J H-H = 6.6 Hz, 1H), 3.583.54 (m, 1H), 3.18-3.15 (m, 2H), 3.10-3.08 (m, 1H), 2.95-2.90 (m, 2H), 2.77 (d, 3J H-H =
12.6 Hz, 1H), 2.20 (s, 3H), 2.12-2.07 (m, 2H), 2.09 (s, 3H), 1.44 (d, 3J P-H = 15.6 Hz, 9H),
1.34 (d, 3J P-H = 15.6 Hz, 9H); 13C{1H} NMR (CDCl 3 ): δ 164.7 (d, 3J P-C = 6.2 Hz), 157.5,
153.3 (d, 2J P-C = 11.9 Hz), 140.2 (d, 1J P-C = 15.8 Hz), 129.2, 128.9, 127.3, 127.1, 126.9,
121.8 (d, 2J P-C = 5.4 Hz), 88.7 (d, 2J P-C = 26.5 Hz), 53.1 (d, 2J P-C = 2.7 Hz), 41.1 (d, 3J P-C
= 2.1 Hz), 38.7 (d, 2J P-C = 10.0 Hz), 38.6 (d, 1J P-C = 15.8 Hz), 38.3 (d, 1J P-C = 16.0 Hz),
31.0 (d, 2J P-C = 6.2 Hz), 30.7 (d, 2J P-C = 6.5 Hz), 24.0 (d, 3J P-C = 3.6 Hz), 18.8; 31P{1H}
NMR (CDCl 3 ): δ 59.48; IR: 2926 (s), 2854 (s), 2719 (w), 1612 (w), 1566 (w), 1457 (s),
1379 (s), 1270 (w), 1140 (w), 1083 (w), 1026 (w), 720 (w), 637 (m) cm-1. Anal. Calcd for
C 26 H 39 F 3 NO 3 PPdS: C, 48.79; H, 6.14; N, 2.19. Found: C, 48.88; H, 6.07; N, 2.33.
[(2-Diphenylphosphinocyclohexene-1-(tert-butyl)imine)Pd(allyl)]OTf (6Pd):
light brown solid (0.340 g, 85.1%); mp 160 oC dec; 1H NMR (CDCl 3 ): δ 7.65 (d, 4J P-H =
3.6 Hz, 1H), 7.51 (br s, 6H), 7.34-7.33 (m, 4H), 5.77-5.70 (m, 1H), 4.90-4.87 (m, 1H),
3.89-3.85 (m, 1H), 3.39 (s, 1H), 2.96 (d, 3J P-H = 10.8 Hz, 1H), 2.56 (s, 2H), 1.85-1.81 (m,
4H), 1.73-1.64 (m, 2H), 1.14 (s, 9H);
13
C{1H} NMR (CDCl 3 ): δ 163.9 (d, 3J P-C = 11.5
Hz), 148.8 (d, 2J P-C = 12.2 Hz), 133.3, 132.1 (d, 4J P-C = 31.5 Hz), 129.8 (d, 2J P-C = 19.6
Hz), 129.3 (d, 1J P-C = 32.9 Hz), 127.8 (d, 1J P-C = 45.7 Hz), 121.0 (d, 2J PC = 6.5 Hz), 80.7
(d, 2J P-C = 31.1 Hz), 64.3, 56.9 (d, 2J P-C = 4.8 Hz), 31.6 (d, 2J P-C = 9.8 Hz), 30.4, 29.9,
22.5 (d, 3J P-C = 4.5 Hz), 21.5;
31
P{1H} NMR (CDCl 3 ): δ 31.78; IR: 2916 (s), 2854 (s),
44
2719 (m), 2667 (m), 1628 (w), 1586 (w), 1457 (s), 1379 (s), 1259 (s), 1145 (m), 1099
(m), 1026 (m), 974 (m), 912 (m), 798 (m), 720 (m), 637 (m) cm-1. Anal. Calcd for
C 27 H 33 F 3 NO 3 PPdS•1/2(C 4 H 8 O): C, 51.07; H, 5.47; N, 2.05. Found: C, 51.14; H, 5.44;
N, 2.08.
[(2-Di-tert-butylphosphinocyclohexene-1-(2,6-xylyl)imine)Pd(allyl)]OTf (7Pd):
dark brown solid (0.483 g, 36.5%); mp 155-160 oC dec; 1H NMR (CDCl 3 ): δ 7.65 (s,
1H), 7.15-7.08 (m, 3H), 5.69-5.64 (m, 1H), 4.13 (d, 3J H-H = 6.6 Hz, 1H), 3.54-3.50 (m,
1H), 3.12-3.09 (m, 1H), 2.89-2.86 (m, 1H), 2.79 (d, 3J H-H = 12.6 Hz, 1H), 2.75-2.72 (m,
1H), 2.65-2.56 (m, 2H), 2.22 (s, 3H), 2.12 (s, 3H), 1.87-1.81 (m, 4H), 1.49 (d, 3J P-H =
15.6 Hz, 9H), 1.39 (d, 3J P-H = 15.0 Hz, 9H); 13C{1H} NMR (CDCl 3 ): δ 169.0 (d, 3J P-C =
8.6 Hz), 158.1, 146.7 (d, 2J P-C = 7.8 Hz), 139.4 (d, 1J P-C = 13.1 Hz), 129.4, 129.0, 127.3,
127.1, 126.8, 122.0 (d, 2J P-C = 5.4 Hz), 88.0 (d, 2J P-C = 26.2 Hz), 55.1 (d, 2J P-C = 3.9 Hz),
39.5 (d, 1J P-C = 13.1 Hz), 38.8 (d, 1J P-C = 13.9 Hz), 35.7 (d, 2J P-C = 8.7 Hz), 32.9 (d, 3J P-C
= 2.9 Hz), 31.8 (d, 2J P-C = 6.6 Hz), 31.4 (d, 2J P-C = 6.9 Hz), 21.8 (d, 3J P-C = 2.3 Hz), 21.4
(d, 4J P-C = 1.2 Hz), 18.9; 31P{1H} NMR (CDCl 3 ): δ 71.63; IR: 2916 (s), 2854 (s), 1618
(w), 1560 (w), 1457 (s), 1379 (s), 1270 (s), 1223 (m), 1140 (m), 1031 (m), 912 (w), 798
(w), 720 (w), 637 (m) cm-1. Anal. Calcd for C 27 H 41 F 3 NO 3 PPdS: C, 49.58; H, 6.32; N,
2.14. Found: C, 49.58; H, 6.37; N, 2.16.
[(2-Di-tert-butylphosphinocyclohexene-1-(tert-butyl)imine)Pd(allyl)]OTf (8Pd):
green-brown solid (0.706 g, 53.0%); mp 105 oC dec; 1H NMR (CDCl 3 ): δ 7.85 (d, 4J P-H =
3.0 Hz, 1H), 5.64-5.57 (m, 1H), 5.08-5.05 (m, 1H), 3.91 (d, 3J H-H = 7.2 Hz, 1H), 3.79-
45
3.76 (m, 1H), 2.82 (d, 3J H-H = 12.0 Hz, 1H), 2.57-2.56 (m, 4H), 1.78-1.67 (m, 4H), 1.45
(s, 9H), 1.45 (d, 3J P-H = 14.4 Hz, 9H), 1.36 (d, 3J P-H = 14.4 Hz, 9H);
13
C{1H} NMR
(CDCl 3 ): δ 166.9 (d, 3J P-C = 9.2 Hz), 149.5 (d, 2J P-C = 9.8 Hz), 132.4 (d, 1J P-C = 12.8 Hz),
119.6 (d, 2J P-C = 5.6 Hz), 82.4 (d, 2J P-C = 26.7 Hz), 65.4, 56.6, 38.6 (d, 1J P-C = 10.9 Hz),
38.6 (d, 1J P-C = 11.3 Hz), 34.4 (d, 2J P-C = 9.5 Hz), 32.4 (d, 3J P-C = 2.1 Hz), 31.9 (d, 2J P-C =
6.2 Hz), 31.7 (d, 2J P-C = 6.5 Hz), 30.7, 22.7 (d, 3J P-C = 2.3 Hz), 21.5;
31
P{1H} NMR
(CDCl 3 ): δ 75.37; IR: 2926 (s), 2854 (s), 1462 (s), 1379 (m), 1265 (m), 1145 (w), 1031
(w), 798 (w), 720 (w), 637 (m) cm-1. Anal. Calcd for C 23 H 41 F 3 NO 3 PPdS: C, 45.59; H,
6.82; N, 2.31. Found: C, 45.65; H, 6.85; N, 2.51.
[(2-Di-tert-butylphosphinocycloheptene-1-(tert-butyl)imine)Pd(allyl)]OTf (9Pd):
light yellow solid (0.322 g, 62.0%); mp 149-154 oC dec; 1H NMR (CDCl 3 ): δ 8.07 (d,
4
J P-H = 2.4 Hz, 1H), 5.63-5.57 (m, 1H), 5.22-5.19 (m, 1H), 3.93 (d, 3J H-H = 7.2 Hz, 1H),
3.89-3.85 (m, 1H), 2.98-2.88 (m, 2H), 2.79-2.68 (m, 3H), 1.92-1.88 (m, 1H), 1.86-1.81
(m, 1H), 1.78-1.72 (m, 2H), 1.55-1.48 (m, 2H), 1.45 (s, 9H), 1.43 (d, 3J P-H = 14.4 Hz,
9H), 1.33 (d, 3J P-H = 15.0 Hz, 9H);
13
C{1H} NMR (CDCl 3 ): δ 167.8 (d, 3J P-C = 10.0 Hz),
156.8 (d, 2J P-C = 9.8 Hz), 156.1 (d, 1J P-C = 13.7 Hz), 119.7 (d, 2J P-C = 5.4 Hz), 85.1 (d,
2
J P-C = 26.1 Hz), 65.7, 54.2 (d, 2J P-C = 4.1 Hz), 39.4 (d, 1J P-C = 13.3 Hz), 38.8 (d, 1J P-C =
10.9 Hz), 38.6 (d, 2J P-C = 11.6 Hz), 34.5 (d, 3J P-C = 2.0 Hz), 31.6, 31.61 (d, 2J P-C = 6.2
Hz), 31.3 (d, 2J P-C = 6.8 Hz), 30.9, 27.5 (d, 3J P-C = 2.4 Hz), 25.2 (d, 4J P-C = 2.0 Hz);
31
P{1H} NMR (CDCl 3 ): δ 80.64; IR: 2937 (s), 2854 (s), 1623 (w), 1545 (w), 1462 (s),
1374 (s), 1265 (s), 1223 (m), 1145 (m), 1031 (m), 964 (w), 922 (w), 865 (w), 803 (w),
46
751 (w), 720 (w), 637 (s) cm-1. Anal. Calcd for C 24 H 43 F 3 NO 3 PPdS: C, 46.49; H, 7.00;
N, 2.26. Found: C, 46.58; H, 7.00; N, 2.27.
[(2-Di-tert-butylphosphinocyclobutene-1-(tert-butyl)imine)Pd(allyl)]OTf (10Pd):
Sticky brown solid (0.415 g, 71.4%); 1H NMR (CDCl 3 ): δ 7.94 (d, 4J P-H = 1.8 Hz, 1H),
5.60-5.58 (m, 1H), 5.38-5.35 (m, 1H), 4.19-4.16 (m, 1H), 3.99 (d, 3J H-H = 6.6 Hz, 1H),
3.13-3.02 (m, 4H), 2.54 (d, 3J H-H = 12.0 Hz, 1H), 1.42 (d, 3J P-H = 15.0 Hz, 9H), 1.42 (s,
9H), 1.26 (d, 3J P-H = 15.0 Hz, 9H); 13C{1H} NMR (CDCl 3 ): δ 160.3 (d, 1J P-C = 10.7 Hz),
157.5 (d, 3J P-C = 3.9 Hz), 140.5 (d, 2J P-C = 8.6 Hz), 118.5 (d, 2J P-C = 5.3 Hz), 92.3 (d, 2J PC
= 25.6 Hz), 65.1, 45.2, 39.9 (d, 1J P-C = 14.6 Hz), 37.6 (d, 1J P-C = 19.2 Hz), 35.3 (d, 3J P-C
= 5.3 Hz), 32.1 (d, 2J P-C = 16.6 Hz), 30.6 (d, 2J P-C = 5.7 Hz), 30.5, 30.3 (d, 2J P-C = 4.5
Hz); 31P{1H} NMR (CDCl 3 ): δ 55.18; IR: 2916 (s), 2854 (s), 2719 (m), 2293 (w), 1602
(w), 1457 (s), 1374 (s), 1259 (s), 1151 (m), 1088 (m), 1026 (s), 933 (m), 876 (w), 803
(m), 720 (m), 632 (m) cm-1. Anal. Calcd for C 21 H 37 F 3 NO 3 PPdS: C, 43.64; H, 6.45, N,
2.42. Found: C, 43.00; H, 6.26; N, 2.21.
2.5 Crystallography of 1Pd, 8Pd, and 9Pd
A summary of crystal data and collection parameters for crystal structures of 1Pd,
8Pd, and 9Pd are provided in Table 2.3. Detailed descriptions of data collection, as well
as data solution, are provided below. ORTEP diagrams were generated with the Diamond
3 software package.96 For each sample, a suitable crystal was mounted on a glass fiber or
polymer loop using Paratone-N hydrocarbon oil. The crystal was transferred to a Siemens
SMART97 or Apex 2 diffractometer with a CCD area detector, centered in the X-ray
47
beam, and cooled to 140 K using a nitrogen-flow low-temperature apparatus that had
been previously calibrated by a thermocouple placed at the same position as the crystal.
An arbitrary hemisphere of data was collected using 0.3° ω scans, and the data were
integrated by the program SAINT.98 The final unit cell parameters were determined by a
least-squares refinement of the reflections with I > 2σ(I). Data analysis using Siemens
XPREP and the successful solution and refinement of the structure determined the space
group.99 Empirical absorption corrections were applied using the program SADABS.100
Equivalent reflections were averaged, and the structures were solved by direct methods
using the SHELXTL software package.101 Unless otherwise noted, all non-hydrogen
atoms were refined anisotropically.
1Pd: X-ray quality crystals were grown from a pentane layered THF solution at 25 °C. There were two molecules of 1Pd in the asymmetric unit. One triflate anion was
disordered over two positions in a nearly 1:1 ratio. The central carbons of both allyl
moieties in the asymmetric unit were also disordered over two positions each (3:1 and 3:2
ratios). The final cycle of full-matrix least-squares refinement was based on 11637
observed reflections and 796 variable parameters and converged yielding final residuals:
R obs = 0.0432, R all = 0.0595, and GOF = 1.014.
8Pd: X-ray quality crystals were grown from a pentane-layered THF solution at 25 °C. One tert-butyl group was disordered over two positions in a 1:1 ratio. Its carbon
atoms were modeled isotropically. The final cycle of full-matrix least-squares refinement
was based on 4754 observed reflections and 296 variable parameters and converged
yielding final residuals: R obs = 0.0696, R all = 0.0760, and GOF = 1.172.
48
9Pd: X-ray quality crystals were grown from a pentane-layered THF solution at 25 °C. The crystals were twinned about a C 2 rotation axis bisecting a and b (Twin Law:
001 0-10 100). Additionally, one triflate anion was disordered over two positions in a 3:2
ratio. Despite these complications, all non-hydrogen atoms were still refined
anisotropically. The final cycle of full-matrix least-squares refinement was based on
12351 observed reflections and 686 variable parameters and converged yielding final
residuals: R obs = 0.0484, R all = 0.0574, and GOF = 1.069.
49
Table 2.3 Crystallographic data for compounds 1Pd, 8Pd, and 9Pd.
Compound
1Pd
8Pd
9Pd
Formula
C 30 H 31 F 3 NO 3 PPdS
C 23 H 41 F 3 NO 3 PPdS
C 24 H 43 F 3 NO 3 PPdS
Formula weight
679.99
606.00
620.02
Space group
P2 1 /c
P-1
P2 1 /n
Crystal System
Monoclinic
Triclinic
Monoclinic
Temperature (K)
140
140
140
a (Å)
21.331(4)
10.315(2)
22.407(3)
b (Å)
9.6895(17)
10.453(2)
11.3067(16)
c (Å)
28.509(5)
13.040(3)
22.479(3)
α (°)
90.00
73.83(3)
90.00
β (°)
98.402(4)
79.74(3)
91.5780(10)
γ (°)
90.00
84.99(3)
90.00
5829.3(17)
1327.8(5)
5692.9(14)
8
2
8
Density calc (g/cm )
1.550
1.516
1.447
Diffractometer
Siemens SMART
Bruker APEX2
Bruker APEX2
Radiation
Mo-K α (λ = 0.71073 Å)
Mo-K α (λ = 0.71073 Å)
Mo-K α (λ = 0.71073 Å)
Monochromator
Graphite
Graphite
Graphite
Detector
CCD detector
CCD detector
CCD detector
Scan type, width
ω, 0.3°
ω, 0.3°
ω, 0.3°
Scan speed (s)
20
10
10
Reflections measured
Hemisphere
Hemisphere
Hemisphere
2θ range (°)
1.92-56.68
3.30-52.12
3.60-56.06
Crystal dimensions (mm)
0.20 x 0.18 x 0.12
0.40 x 0.05 x 0.04
0.40 x 0.08 x 0.08
Reflections measured
64333
30580
76098
Unique reflections
14493
5201
13680
Observations (I > 2σ(I))
11637
4754
12351
R int
0.0643
0.0400
0.0516
Parameters
796
296
686
R obs , R w , R all
0.0432, 0.1002, 0.0595
0.0696, 0.1881, 0.0760
GoF
1.014
1.172
3
V (Å )
Z
3
50
0.0484, 0.1296, 0.0574
1.069
Chapter 3
Further Modification of the 3Iminophosphine-Based Palladium
Catalyst
3.1 Introduction
Expanding upon the results of the second chapter, investigation of the steric and
electronic properties of the 3IP ligands and their resulting effect on the catalytic
efficiency of their respective palladium complexes continued. This led to the synthetic
development and characterization of four new 3IP ligands and [(3IP)Pd(allyl)]OTf
complexes. Understanding that changes made to each of the three tunable domains can
either promote or destroy hydroamination catalysis with these complexes, further
alteration to these three domains proceeded. Two of the new 3IP ligands (11 and 12)
contained alterations to the phosphine moiety, while the other two each varied the
basicity of the imine moiety (13) and the composition of the unsaturated backbone (14)
(Figure 3-1). Ligands 11 and 12 each started with the framework of ligand 4, but
exchanged the tertiary carbons of the tert-butyl phosphine groups for the secondary
51
carbons of cyclohexyl groups and primary carbons of iso-butyl groups, respectively.
Figure 3-1 3-Iminophosphine (3IP) ligands 11-14.
It was previously noted that ligands utilizing alkyl groups instead of aryl groups on the
phosphine significantly increased catalytic activity of their palladium complexes,102 so
naturally variation of the alkyl group substitution was thought to impact the catalysis. The
more basic tert-butyl imine moiety of 2Pd, 4Pd, and 10Pd also led to significantly
greater catalytic activity than the less basic 2,6-xylyl imine moieties of 1Pd, 3Pd, 5Pd,
and 7Pd, leading to the synthesis of 13, with an even more basic dimethylamino group on
the imine. The last ligand in this ancillary array was generated from the notion that
decreasing size of the alicyclic backbone led to increased turnover frequency in
[(3IP)Pd(allyl)]OTf complexes. Ligand 14 was synthesized without an alicyclic
backbone, instead replacing it with a hydrogen atom and tert-butyl group at the 1 and 2
positions, respectively. All ligands in this array were coordinated to Pd(allyl)Cl and the
chlorine abstracted with AgOTf to produce their respective [(3IP)Pd(allyl)]OTf
complexes, 11Pd-14Pd. The [(3IP)Pd(allyl)]OTf complexes were then screened for their
52
potential to catalyze the hydroamination of 1,1-dimethylallene with substituted anilines in
a manner analogous to that of the previous chapter.
3.2 Results and Discussion
Ligands 12 and 13 were prepared in a manner analogous to that described by
Zingales et al.,102 while 11 and 14 required modified methodologies in order to ensure
reaction completion and selectivity for the desired products. Due to its lack of appreciable
solubility in diethylether, lithium dicyclohexylphosphide required dissolution in THF in
order to be added in a dropwise manner to 2-chlorocyclopentene-1-(tert-butyl)imine.
These conditions produced multiple phosphorus containing products, most likely due to
the highly reactive nature of this phosphide when compared to lithium di-tertbutylphosphide. A methodology analogous to Kuchenbeiser et al.,71 utilizing a lower
reaction temperature (-78 oC), a solvent in which the phosphide was not readily soluble
(toluene), a longer reaction time (10 h), and a celite padded frit for extraction, was
employed in order to produce the desired product in good yield and purity.
Ligand 14, with its lack of alicyclic backbone, was open to two different
diastereomers upon attack of the phosphide at the 2 position. Because of this, a
methodology analogous to Shaffer et al. was employed in order to select the cis addition
product over that of the trans, as the stereochemistry of the trans isomer does not permit
chelation to a metal center.47 A dilute solution of chloroimine in pentane was added to a
slurry of lithium di-tert-butylphosphide in pentane at -78 oC, allowed to slowly warm to
room temperature, then heated at 50 oC for 10 hours to ensure reaction completion. After
cooling to room temperature ligand 14 was extracted with pentane using a celite padded
53
frit. Complexes 11Pd and 13Pd required recrystallization of their respective
(3IP)Pd(allyl)Cl complexes from pentane in order to achieve successful halide abstraction
with AgOTf in the final step.
As noted in the previous chapter, silver chloride produced during chloride
removal from (3IP)Pd(allyl)Cl complexes was readily coordinated by excess 3IP ligand.
This was first observed when an unidentified crystalline material was found to be much
less soluble in THF than 4Pd, both of which had been co-crystallized from a pentane
layered THF solution. When the low solubility material was observed by 1H NMR
spectroscopy, all resonances seemed to indicate uncoordinated ligand, yet the physical
appearance of the material seemed to indicate some type of metal complex, as free ligand
4 is a viscous liquid at room temperature. Analysis via 31P NMR spectroscopy displayed
neither resonances of 4 nor 4Pd of significant intensity, but instead showed two doublets
with the same chemical shift. The compound was further characterized by X-ray
crystallography, which revealed the isolated material to be a silver chloride complex
(Figure 3-2).103 This was a dimer of AgCl and monodentate 4, coordinated only through
the phosphine (4AgCl), which had co-crystallized with 4Pd. To ensure that this complex
was not indeed the active hydroamination catalyst, the compound was screened similar to
the other species and displayed negligible catalytic activity for the hydroamination of 1,1dimethylallene with 4-fluoroaniline, activity that was consistent with the trace remaining
4Pd in the crystalline mixture.
54
Figure 3-2 ORTEP (50% thermal ellipsoids) of 4AgCl. Hydrogen atoms have been
omitted for clarity. Selected bond lengths (in Å): Ag1-P1 = 2.3793(9), Ag1-Cl1 =
2.478(1), Ag1-Cl2 = 2.574(1), Ag1-Ag2 = 3.3422(4), Ag2-P2 = 2.381(1), Ag2-Cl1 =
2.508(1), Ag2-Cl2 = 2.542(1). Bond angles (in deg): Ag1-Ag2-P2 = 175.32(3), Ag2Ag1-P1 = 169.32(3), Cl1-Ag1-Cl2 = 96.70(3), Cl1-Ag2-Cl2 = 96.77(3), Ag1-Cl1-Ag2
= 84.17(3), Ag1-Cl2-Ag2 = 81.58(3), Cl1-Ag1-P1 = 140.30(4), Cl2-Ag2-P2 =
126.38(3).
All of the ligands and palladium complexes described in this chapter are
diamagnetic, and so an analysis of their NMR spectroscopic features proves quite
insightful. Similar to the original array of 10 3IP ligands from Chapter 2, substantial
differences were noted between
1
H,
13
C, and
31
P NMR spectra of the free 3-
iminophosphine ligands and those of the [(3IP)Pd(allyl)]OTf complexes. These trends are
summarized in Table 3.1 below. As was the case with all other 3IP palladium complexes
synthesized by our group, downfield shifts in the 31P NMR signals were observed upon
coordination of the ligand to the palladium, with a further downfield shift upon chloride
abstraction for ligands 11-14. The change in this shift between free ligand and complex
(Δ31P δ) is far less varied than the array of ten ligands and complexes found in the
55
previous chapter, since all 3IP ligands in this array have alkyl groups on the phosphine
moiety. As larger Δ31P δ seems to denote tighter binding of the phosphine to the metal
center, it is implied that 12 and 14, both displaying more than 10 ppm larger difference in
their chemical shifts than 11 and 13, are most strongly bound to palladium in this array.
Table 3.1 Comparison between selected 31P and 1H NMR resonances of
[(3IP)Pd(allyl)]OTf complexes and their respective free 3IP ligands (all values in
ppm).
3IPPda
3IPPda
3IPb
Δ31P δ
3IPb
Δ1H δ
31
31
R'N=CH 1H δ R'N=CH 1H δ
Pδ
Pδ
30.20
-20.57
50.77
8.05
8.73
-0.68
11
5.60
-56.13
61.73
8.15
8.79
-0.64
12
60.11
12.93
47.18
7.32
7.92
-0.60
13
88.67
23.09
65.58
8.28
8.12
0.16
14
a
3IPPd refers to [(3IP)Pd(allyl)]OTf complex. b 3IP refers to uncoordinated ligand.
In fact, 12 and 14 display the greatest Δ31P δ values of all 14 ligands in this thesis, with
values of 61.73 and 65.58 ppm, respectively. Both 11 and 12 and their respective
palladium complexes displayed 1H NMR resonances for their imine C-H similar to
ligands and complexes from the previous chapter, while 13 and 14 showed
uncharacteristic variations from the norm. Whereas free ligand imine C-H tends to rest at
or around 9 ppm, with an upfield shift (denoting loss of double bond character of the
imine moiety upon coordination of the imine nitrogen to palladium) to about 8 ppm for
the respective 3IP palladium complexes, 13 starts with a resonance of 7.92 ppm that
shifts upfield to 7.32 ppm for 13Pd. The change in imine C-H resonance upon
coordination (Δ1H δ) is still comparable to those of the other alkyl phosphino-complexes,
-0.60 ppm (the negative number denoting an upfield shift from ligand to triflated
complex). Ligand 14, however, displayed a reversed value for Δ1H δ, with a 0.16 ppm
downfield shift between free ligand and [(3IP)Pd(allyl)]OTf complex. This may imply
56
that the conjugated pi system of the non-cyclic alkene is permitting the electronegative
phosphorus atom to draw electron density away from the imine carbon and hydrogen.
This is most likely a result of significant electron donation from the phosphorus atom to
the metal center.
Similar to the complexes of the previous chapter, the trends in the
13
C NMR
spectra parallel those of the 1H NMR spectra, as there are significant changes in the
chemical shift of the imine carbon between free and coordinated ligands. The terminal
carbons of the allyl ligands (cis and trans to phosphorus) were readily differentiated
based upon both the magnitude of the J P-C coupling constant, and their relative chemical
shift values. The trans carbon was typically located downfield from the cis carbon and
displayed a J P-C coupling constant an order of magnitude larger, if the cis carbon
displayed coupling to phosphorus at all.
Results for the catalytic screening of compounds 11Pd-14Pd, along with
compounds 2Pd, 4Pd, and 10Pd for comparison, are summarized in Table 3.2 below. All
four complexes displayed greatly decreased catalytic activity in the hydroamination of
1,1-dimethylallene with substituted anilines when compared to the most active complexes
from the previous array. Changing from the tertiary carbons of the phosphine moiety of
4Pd to the secondary carbons of 11Pd and the primary carbons of 12Pd showed a
marked drop in activity for each successive decrease in substitution. The phenyl groups
of 2Pd seem to afford it intermediate catalytic activity when compared to 4Pd and 12Pd.
The cone angle of these phosphines decreases going from tert-butyl to cyclohexyl to
phenyl to iso-butyl groups,104 indicating that steric bulk of the phosphine substituents
may play an important role in hydroamination catalysis.
57
Table 3.2 Conversion to product A for the [(3IP)Pd(allyl)]OTf catalyzed
hydroamination of 1,1-dimethylallene with aryl amines (complexes 11Pd-14Pd).
Conversion was monitored via 1H NMR spectroscopy.
Aniline
2Pd
4Pd
10Pd
11Pd
12Pd
13Pd
14Pd
--
60%
91%
7%
--
18%
16%
--
--
NP
NP
12%
--
NP
NP
9%
--
NP
NP
8%
--
NP
7%
6%
--
14%
NP
9%
--
NP
NP
2-Methyl
---Aniline
3-Methyl
13%
>95% 92%
Aniline
4-Methyl
16%
>95% >95%
Aniline
4-tertButyl 17%
88%
>95%
Aniline
4-Fluoro
9%
65%* 90%
Aniline
3-Methoxy 12%
62%
72%
Aniline
* indicates reaction at 70oC. “--” indicates
performed.
<5%. NP indicates a trial that was not
Substitution of the more basic dimethylamino imine for the tert-butyl
imine
of
4Pd to make 13Pd did not increase its hydroamination catalytic activity. This may be
attributed to tighter binding of its imine nitrogen donor atom. Stronger coordination of
the nitrogen decreases the hemilability of this ligand more than the other complexes,
restricting coordination of the substrate amine during the proposed catalytic cycle
(Scheme 1.4). Removal of the alicyclic backbone of 4Pd to make 14Pd also did not
increase catalytic activity. The extremely electron donating nature of its phosphine
moiety compared to the other palladium complexes in this thesis (based upon its greater
58
Δ31P δ value) may create a much less Lewis acidic metal center that is less likely to be
coordinated by substrates for hydroamination. Also, the lack of a cyclic backbone
eliminates the angular strain of this ligand domain, a strain that is maximized in the most
active catalyst thus far, 10Pd.
3.3 Conclusions
Continued efforts to improve the performance of 3-iminophosphine-based palladium
catalysts for the hydroamination of 1,1-dimethylallene with substituted anilines have
concluded that the ligand composed of di-tert-butyl phosphine, cyclobutene backbone,
and tert-butyl imine still provides the most active palladium catalyst for this
hydroamination when compared to the other complexes in this collection. Tertiary
aliphatic groups on the phosphine are necessary for effective catalysis, as primary and
secondary aliphatics and aromatic groups decrease the catalytic activity. Imine moieties
of both low and high basicity were also shown to hinder catalysis, indicating that
intermediate basicity is ideal in this system. Increased ring strain in the alicyclic
backbone was found to have beneficial effects for hydroamination catalysis, whereas
removal of this domain was shown to slow catalysis in all cases.
3.4 Experimental Section
3.4.1 General Methods and Instrumentation:
Alicyclic α,β-unsaturated β-chloroaldehydes and β-chloroimines were handled
under ambient atmospheric conditions. All other manipulations were performed under an
inert N 2 atmosphere using standard Schlenk and drybox techniques. Solvents were dried
59
prior to use; methylene chloride was passed through two columns of 4 Å molecular sieves
and degassed with nitrogen. Pentane, diethyl ether, and toluene were passed through
columns of activated alumina and 4 Å molecular sieves and degassed with nitrogen.
Tetrahydrofuran was distilled from sodium metal and degassed with nitrogen. nButyllithium (1.6 M in hexanes), (allyl)palladium(II) chloride dimer, di-isobutylphosphine, di-tert-butylchlorophosphine, lithium aluminum hydride, and silver
triflate
were
purchased
from Strem and
used
without
further
purification.
Dicyclohexylphosphine was purchased commercially and used without further
purification. Phosphorus oxychloride, tert-butylamine, pinacolone, and cyclopentanone
were purchased from Acros and used without further purification. 1,1-Dimethylallene
was
purchased
from
Alfa
Aesar
and
used
without
further
purification.
Dimethylformamide was purchased from BDH and stored over 4 Å molecular sieves.
N,N-Dimethylhydrazine was purchased from Sigma-Aldrich and used without further
purification. Anilines were also purchased from Sigma-Aldrich or another commercial
source and dried over calcium hydride, either neat (liquid anilines) or as solutions in
methylene chloride (solid anilines). Liquid anilines were freeze-pump-thawed three times
and vacuum distilled. Solutions of solid anilines in methylene chloride were freezepump-thawed three times, filtered, and the methylene chloride removed via reduced
pressure. CDCl 3 was purchased from Cambridge Isotope Laboratories, vacuum
transferred from CaH 2 , and stored over 4 Å molecular sieves. Benzene-d 6 was also
purchased from Cambridge Isotope Laboratories, vacuum transferred from sodium metal,
and stored over 4 Å molecular sieves. Silica gel (Porosity: 60 Å, Particle size: 40-63 μm)
was purchased from Sorbent Technologies and used as received. 1H and
60
13
C NMR data
were obtained on a 600 MHz Inova or 400 MHz VXRS NMR spectrometer at ambient
temperature at 599.9 MHz for 1H NMR and 150.8 MHz for 13C NMR and 399.95 MHz
for 1H NMR and 100.56 MHz for
13
C NMR, respectively. All
31
P NMR spectra were
collected on a 400 MHz VXRS NMR spectrometer at ambient temperature at 161.90
MHz. All spectra were taken using C 6 D 6 or CDCl 3 as the NMR solvent. 1H NMR shifts
are given relative to the residual solvent resonances at 7.16 and 7.26 ppm, respectively,
and 13C NMR shifts are given relative to the residual solvent peak of CDCl 3 (77.36 ppm).
31
P NMR spectra were externally referenced to 0.00 ppm with 5% H 3 PO 4 in D 2 O. IR
samples were prepared as Nujol mulls and taken between KBr plates on a Perkin-Elmer
XTL FTIR spectrophotometer. Melting points were observed on a capillary melting point
(Uni-Melt) apparatus in sealed capillary tubes and are uncorrected. X-ray structure
determinations were performed at the Ohio Crystallographic Consortium, housed at The
University of Toledo. Elemental analyses were determined by Atlantic Microlab, Inc.,
Norcross, GA or Galbraith Laboratories, Inc., Knoxville, TN. High resolution mass
spectrometry using electrospray ionization was performed at the University of Illinois
Mass Spectrometry Laboratory, Urbana, IL.
3.4.2 General Procedure for the Catalytic Hydroamination Screening of Compounds
11Pd-14Pd
All manipulations were performed under an N 2 atmosphere. 3-Methyl-1,2-butadiene (68
mg, 1 mmol) was added to a mixture of amine (0.5 mmol), [(3IP)Pd(allyl)]OTf complex
(5 mol%), and deuterated benzene (0.8 ml). Conversion to products was monitored via 1H
NMR spectroscopy. Hydroamination products formed were reported previously.3
61
3.4.3 Synthesis and Characterization of Reaction Products
The following compounds were synthesized as previously reported: LiPtBu 2 ,3 2chlorocyclopentenecarboxaldehyde,88 2-chlorocyclopentene-1-(tert-butyl)imine,88 Z-3chloro-3-tert-butylpropenal,105 and E-2-chloro-2-tert-butylethene-1-(tert-butyl)imine.105
LiPiBu 2 and LiPCy 2 were both synthesized using methodologies analogous to Shaffer et
al.88
2-Chlorocyclopentene-1-(dimethylamino)imine:
golden yellow liquid (2.817 g, 62.46%); 1H NMR (CDCl 3 ): δ 7.17 (s, 1H), 2.90 (s, 6H),
2.67-2.64 (m, 2H), 2.63-2.60 (m, 2H), 1.97-1.92 (m, 2H);
13
C{1H} NMR (CDCl 3 ): δ
134.8, 128.7, 128.6, 43.0, 39.0, 31.1, 21.3; IR: 2956 (s), 2857 (s), 2790 (s), 2592 (w),
2493 (w), 2393 (w), 2305 (w), 2106 (w), 1893 (w), 1761 (w), 1628 (m), 1551 (s), 1469
(s), 1441 (s), 1402 (m), 1364 (m), 1325 (s), 1270 (s), 1198 (w), 1132 (s), 1104 (s), 1044
(s), 944 (s), 911 (m), 878 (m), 851 (m), 801 (m), 751 (m), 553 (s) cm-1.
2-Dicyclohexylphosphinocyclopentene-1-(tert-butyl)imine (11):
To a Schlenk tube was added lithium dicyclohexylphosphide (1.6 equiv.), a stirbar, and
toluene (30 ml). This rapidly stirring slurry was cooled to -78 oC via a dry ice/isopropanol
bath for 10 min before slowly adding a solution of degassed 2-chlorocyclopentene-1(tert-butyl)imine (1.00 equiv.) in toluene (30 ml), also cooled to -78 oC for 10 min. The
reaction mixture was allowed to slowly warm to room temperature and react for 10
additional hours before solvent was removed in vacuo. The resulting oil was triturated
62
with pentane (2 X 10 ml), then extracted into pentane (2 X 20 ml) and passed through a
celite padded frit. Solvent was again removed in vacuo to yield the final product.
thick orange oil (0.615 g, 68.3%); 1H NMR (CDCl 3 ): δ 8.73 (d, 4J P-H = 4.8 Hz, 1H), 2.722.69 (m, 2H), 2.63-2.61 (m, 2H), 1.86-1.58 (m, 14H), 1.30-1.07 (m, 10H), 1.21 (s, 9H);
13
C{1H} NMR (CDCl 3 ): δ 155.3 (d, 2J P-C = 17.9 Hz), 154.2 (d, 3J P-C = 23.7 Hz), 147.2
(d, 1J P-C = 26.8 Hz), 57.6, 37.8 (d, 2J P-C = 6.0 Hz), 33.9 (d, 3J P-C = 5.3 Hz), 33.1 (d, 2J P-C
= 9.5 Hz), 30.9 (d, 1J P-C = 16.3 Hz), 30.7 (d, 2J P-C = 8.1 Hz), 30.3, 27.53 (d, 3J P-C = 0.9
Hz), 27.46 (d, 4J P-C = 3.3 Hz), 26.8 (d, 3J P-C = 0.9 Hz), 23.3 (d, 3J P-C = 0.9 Hz); 31P{1H}
NMR (CDCl 3 ): δ – 20.57; IR: 2926 (s), 2854 (s), 2657 (w), 2262 (w), 1623 (s), 1571 (m),
1446 (s), 1363 (s), 1265 (s), 1213 (s), 1176 (s), 1073 (m), 995 (s), 964 (m), 886 (m), 844
(s), 803 (m), 746 (w), 709 (w) cm-1. HRMS (m/z): [M+H]+ calcd for C 22 H 39 NP 348.2820,
found 348.2828.
[(2-Dicyclohexylphosphinocyclopentene-1-(tert-butyl)imine)Pd(allyl)]OTf (11Pd):
Required
recrystallization
of
(2-dicyclohexylphosphinocyclopentene-1-(tert-
butyl)imine)Pd(allyl)Cl intermediate from pentane before preparation in a manner
analogous to Zingales et al.102
Orange yellow solid (0.155 g, 53.2%); 1H NMR (CDCl 3 ): δ 8.05 (d, 4J P-H = 1.8 Hz, 1H),
5.64-5.57 (m, 1H), 5.15-5.12 (m, 1H), 3.99-3.95 (m, 1H), 3.73 (d, 3J H-H = 7.2 Hz, 1H),
2.92-2.89 (m, 2H), 2.86-2.82 (m, 1H), 2.73-2.70 (m, 1H), 2.49 (d, 3J H-H = 12.0 Hz, 1H),
2.26-2.21 (m, 1H), 2.10-2.01 (m, 3H), 1.88-1.72 (m, 10H), 1.44 (s, 9H), 1.40-1.06 (m,
10H);
13
C{1H} NMR (CDCl 3 ): δ 160.5 (d, 3J P-C = 7.2 Hz), 156.2 (d, 2J P-C = 13.9 Hz),
133.4 (d, 1J P-C = 24.4 Hz), 119.7 (d, 2J P-C = 5.6 Hz), 88.5 (d, 2J P-C = 27.2 Hz), 65.5, 46.4,
63
38.5 (d, 2J P-C = 11.0 Hz), 37.5, 36.1 (d, 1J P-C = 22.9 Hz), 35.3 (d, 1J P-C = 26.1 Hz), 30.5
(d, 2J P-C = 3.9 Hz), 30.3, 29.2, 29.1, 27.2, 27.1 (d, 2J P-C = 3.6 Hz), 27.0 (d, 2J P-C = 5.3
Hz), 26.9 (d, 2J P-C = 2.0 Hz), 26.8, 26.3 (d, 3J P-C = 1.1 Hz), 26.1 (d, 3J P-C = 1.1 Hz), 23.0
(d, 3J P-C = 4.2 Hz);
31
P{1H} NMR (CDCl 3 ): δ 30.20; IR: 2920 (s), 2854 (s), 1460 (s),
1376 (m), 1263 (m), 1146 (w), 1090 (w), 1027 (m), 797 (w), 722 (w), 634 (m) cm-1.
Anal. Calcd for C 26 H 43 F 3 NO 3 PPdS: C, 48.49; H, 6.73; N, 2.17. Found: C, 46.51; H,
6.77; N, 1.90.
2-Di-iso-butyl-phosphinocyclopentene-1-(tert-butyl)imine (12):
Prepared in a manner analogous to that of Zingales et al.102
Red-orange oil (0.718 g, 97.1%); 1H NMR (CDCl 3 ): δ 8.79 (d, 4J P-H = 4.8 Hz, 1H), 2.702.68 (m, 2H), 2.58-2.56 (m, 2H), 1.85-1.82 (m, 2H), 1.60-1.56 (m, 2H), 1.54-1.50 (m,
2H), 1.28-1.24 (m, 2H), 1.21 (s, 9H), 0.96 (d, 3J H-H = 6.6 Hz, 6H), 0.93 (d, 3J H-H = 6.6
Hz, 6H); 13C{1H} NMR (CDCl 3 ): δ 153.54 (d, 2J P-C = 19.2 Hz), 153.53 (d, 3J P-C = 24.4
Hz), 149.7 (d, 1J P-C = 25.0 Hz), 57.6, 36.9 (d, 1J P-C = 9.8 Hz), 34.2 (d, 3J P-C = 5.0 Hz),
34.0 (d, 2J P-C = 6.5 Hz), 30.3, 26.8 (d, 2J P-C = 12.8 Hz), 24.9 (d, 3J P-C = 8.3 Hz), 24.0 (d,
3
J P-C = 10.0 Hz), 22.8 (d, 3J P-C = 1.1 Hz);
31
P{1H} NMR (CDCl 3 ): δ -56.13; IR: 3186
(m), 2958 (s), 2885 (s), 2719 (m), 2657 (w), 2615 (m), 1654 (m), 1623 (s), 1581 (m),
1462 (s), 1415 (m), 1384 (s), 1363 (s), 1332 (m), 1259 (s), 1213 (s), 1166 (s), 1104 (s),
1078 (s), 1042 (s), 964 (m), 922 (m), 896 (m), 834 (m), 803 (s), 735 (m), 704 (m) cm-1.
HRMS (m/z): [M+H]+ calcd for C 18 H 35 NP 296.2507, found 296.2506.
64
[(2-Di-iso-butyl-phosphinocyclopentene-1-(tert-butyl)imine)Pd(allyl)]OTf (12Pd):
Prepared in a manner analogous to that of Zingales et al.102
Tan brown solid (1.208 g, 93.09%); 1H NMR (CDCl 3 ): δ 8.15 (s, 1H), 5.74-5.67 (m, 1H),
5.12-5.10 (m, 1H), 4.09-4.04 (m, 1H), 3.51 (d, 3J H-H = 3.6 Hz, 1H), 2.96-2.93 (m, 2H),
2.79-2.74 (m, 2H), 2.49 (d, 3J H-H = 11.4 Hz, 1H), 2.21-2.16 (m, 1H), 2.11-2.07 (m, 3H),
1.94-1.90 (m, 1H), 1.86-1.84 (m, 1H), 1.73-1.70 (m, 1H), 1.51-1.50 (m, 1H), 1.48 (s,
9H), 1.08 (d, 3J H-H = 6.6 Hz, 3H), 0.98 (d, 3J H-H = 6.6 Hz, 3H), 0.96 (d, 3J H-H = 6.6 Hz,
3H), 0.89 (d, 3J H-H = 6.6 Hz, 3H); 13C{1H} NMR (CDCl 3 ): δ 158.2 (d, 3J P-C = 8.4 Hz),
153.2 (d, 2J P-C = 16.0 Hz), 135.6 (d, 1J P-C = 28.1 Hz), 119.9 (d, 2J P-C = 6.3 Hz), 90.8 (d,
2
J P-C = 28.2 Hz), 66.2, 48.5, 39.6 (d, 2J P-C = 11.5 Hz), 37.5 (d, 1J P-C = 28.7 Hz), 36.3 (d,
1
J P-C = 30.0 Hz), 34.2, 30.4, 26.9, 26.6, 25.0 (d, 3J P-C = 10.3 Hz), 24.4 (d, 3J P-C = 5.0 Hz),
24.3 (d, 3J P-C = 4.0 Hz), 24.2 (d, 3J P-C = 9.5 Hz), 22.6 (d, 3J P-C = 5.1 Hz); 31P{1H} NMR
(CDCl 3 ): δ 5.60; IR: 2916 (s), 2854 (s), 2719 (m), 2677 (w), 1591 (w), 1462 (s), 1379
(m), 1265 (m), 1223 (m), 1140 (m), 1099 (w), 1062 (w), 1031 (m), 948 (w), 844 (w), 808
(w), 751 (w), 720 (w), 637 (m) cm-1. Anal. Calcd for C 22 H 39 F 3 NO 3 PPdS: C, 44.64; H,
6.64; N, 2.37. Found: C, 44.78; H, 6.73; N, 2.39.
2-Di-tert-butylphosphinocyclopentene-1-(dimethylamino)imine (13):
Prepared in a manner analogous to that of Zingales et al.102
orange brown oil (0.320 g, 39.1%); 1H NMR (CDCl 3 ): δ 7.92 (d, 4J P-H = 5.4 Hz, 1H),
2.88 (s, 6H), 2.80-2.77 (m, 2H), 2.65-2.62 (m, 2H), 1.84-1.79 (m, 2H), 1.17 (d, 3J P-H =
12.0 Hz, 18H);
13
C{1H} NMR (CDCl 3 ): δ 154.8 (d, 2J P-C = 22.8 Hz), 137.2 (d, 1J P-C =
65
29.6 Hz), 133.9 (d, 3J P-C = 28.8 Hz), 43.3, 39.3 (d, 3J P-C = 6.5 Hz), 33.4 (d, 2J P-C = 6.6
Hz), 33.0 (d, 1J P-C = 19.3 Hz), 31.1 (d, 2J P-C = 14.3 Hz), 24.2; 31P{1H} NMR (CDCl 3 ): δ
12.93; IR: 2947 (s), 2885 (s), 2854 (s), 2781 (m), 2708 (w), 1628 (w), 1571 (s), 1550 (m),
1534 (m), 1467 (s), 1420 (m), 1384 (m), 1363 (s), 1322 (m), 1265 (s), 1171 (m), 1135 (s),
1104 (s), 1078 (s), 1036 (s), 943 (m), 896 (m), 852 (m), 807 (s), 746 (m), 601 (m), 551 (s)
cm-1.
[(2-Di-tert-butylphosphinocyclopentene-1-(dimethylamino)imine)Pd(allyl)]OTf
(13Pd):
Required
recrystallization
of
(2-di-tert-butylphosphinocyclopentene-1-
(dimethylamino)imine)Pd(allyl)Cl intermediate from pentane before preparation in a
manner analogous to that of Zingales et al.102
sticky yellow solid (0.050g, 63%); 1H NMR (CDCl 3 ): δ 7.32 (s, 1H), 5.86-5.81 (m, 1H),
5.02-4.99 (m, 1H), 4.20-4.16 (m, 1H), 3.90 (d, 3J H-H = 6.0 Hz, 1H), 2.98 (s, 6H), 2.862.77 (m, 4H), 2.74 (d, 3J H-H = 12.0 Hz, 1H), 2.00-1.96 (m, 2H), 1.38 (d, 3J P-H = 15.0 Hz,
9H), 1.25 (d, 3J P-H = 14.4 Hz, 9H); 13C{1H} NMR (CDCl 3 ): δ 153.4 (d, 2J P-C = 13.1 Hz),
141.1 (d, 3J P-C = 2.6 Hz), 128.8 (d, 1J P-C = 19.8 Hz), 120.4 (d, 2J P-C = 4.7 Hz), 91.7 (d,
2
J P-C = 23.5 Hz), 49.9, 45.5, 39.3 (d, 1J P-C = 16.0 Hz), 39.2 (d, 2J P-C = 11.0 Hz), 37.9 (d,
1
J P-C = 17.0 Hz), 30.9 (d, 2J P-C = 6.2 Hz), 30.7 (d, 2J P-C = 5.7 Hz), 30.0 (d, 3J P-C = 5.4
Hz), 23.9 (d, 3J P-C = 3.6 Hz); 31P{1H} NMR (CDCl 3 ): δ 60.11.
66
Z-2-Di-tert-butylphosphino-2-tert-butylethene-1-(tert-butyl)imine (14):
To a Schlenk tube was added lithium di-tert-butylphosphide (1.6 equiv.), a stirbar, and
pentane (50 ml). This rapidly stirring slurry was cooled to -78
o
C via a dry
ice/isopropanol bath for 10 min before slowly adding a solution of degassed E-2-chloro2-tert-butylethene-1-(tert-butyl)imine (1.00 equiv.) in pentane (30 ml), also cooled to -78
o
C. The reaction mixture was allowed to slowly warm to room temperature before heating
at 50 oC for 10 h. The crude product mixture was allowed to cool to room temperature
before passage through a celite padded frit. Solvent was removed in vacuo to yield the
final product.
orange brown oil (0.517 g, 63.2%); 1H NMR (CDCl 3 ): δ 8.12 (d, 3J H-H = 9.2 Hz, 1H),
6.99 (m, 1H), 1.26 (d, 3J P-H = 9.6 Hz, 18H), 1.25-1.21 (br, s, 18H);
13
C{1H} NMR
(CDCl 3 ): δ 160.2, 158.9 (d, 1J P-C = 51.9 Hz), 138.7 (d, 2J P-C = 7.8 Hz), 58.1, 40.7 (d, 1J PC
= 34.4 Hz), 33.7 (d, 2J P-C = 30.8 Hz), 32.8 (d, 2J P-C = 16.2 Hz), 31.9 (d, 3J P-C = 10.8
Hz), 30.4; 31P{1H} NMR (CDCl 3 ): δ 23.09.
[(Z-2-Di-tert-butylphosphino-2-tert-butylethene-1-(tert-butyl)imine)Pd(allyl)]OTf
(14Pd):
Prepared in a manner analogous to that of Zingales et al.102
Light brown solid (0.285g, 58.33%); 1H NMR (CDCl 3 ): δ 8.28-8.27 (m, 1H), 7.32 (dd,
3
J P-H = 39.0 Hz, 3J H-H = 6.0 Hz, 1H), 5.64-5.57 (m, 1H), 4.84-4.83 (m, 1H), 3.92 (d, 3J H-H
= 6.0 Hz, 1H), 3.61-3.57 (m, 1H), 2.94 (d, 3J H-H = 12.6 Hz, 1H), 1.55 (d, 3J P-H = 15.0 Hz,
67
9H), 1.47 (d, 3J P-H = 15.0 Hz, 9H), 1.45 (s, 9H), 1.39 (s, 9H); 31P{1H} NMR (CDCl 3 ): δ
88.67.
(2-Di-tert-butylphosphinocyclopentene-1-(tert-butyl)imine) 2 Ag 2 Cl 2 (4AgCl):
Produced
as
a
byproduct
during
chloride
removal
from
the
(2-di-tert-
butylphosphinocyclopentene-1-(tert-butyl)imine)Pd(allyl)Cl intermediate via addition of
AgOTf when excess 4 was allowed to remain dissolved in the reaction mixture. Work-up
of both complexes simultaneously is achieved via a methodology analogous to that of
Zingales et al.102 Co-crystallization of 4Pd and 4AgCl from a pentane layered THF
solution affords an otherwise pure mixture of both complexes. 4Pd is then readily rinsed
away from 4AgCl with THF (2 × 10 ml).
brown crystalline solid; 1H NMR (CDCl 3 ): δ 9.19 (s, 2H), 2.95-2.92 (m, 4H), 2.81-2.78
(m, 4H), 1.91-1.89 (m, 4H), 1.36 (d, 3J P-H = 15.0 Hz, 36H), 1.28 (s, 18H); 13C{1H} NMR
(CDCl 3 ): δ 158.9 (d, 2J P-C = 9.7 Hz), 151.4 (d, 3J P-C = 17.6 Hz), 135.0 (d, 1J P-C = 14.3
Hz), 59.1, 40.4 (d, 3J P-C = 2.4 Hz), 35.9 (d, 1J P-C = 13.9 Hz), 35.2 (d, 2J P-C = 9.5 Hz), 31.0
(d, 2J P-C = 9.2 Hz), 30.3, 23.5 (d, 3J P-C = 4.2 Hz);
1
31
P{1H} NMR (CDCl 3 ): δ 36.16 (d,
J 107Ag-P = 600.8 Hz), 36.16 (d, 1J 109Ag-P = 689.8 Hz).
3.5 Crystallography of 4AgCl
A summary of crystal data and collection parameters for the crystal structure of
4AgCl is provided in Table 3.3. A detailed description of data collection, as well as the
data solution, is provided below. The ORTEP diagram was generated with the Diamond 3
software package.96 A suitable crystal was mounted on a polymer loop using Paratone-N
hydrocarbon oil. The crystal was transferred to an Apex 2 diffractometer with a CCD
68
area detector, centered in the X-ray beam, and cooled to 140 K using a nitrogen-flow
low-temperature apparatus that had been previously calibrated by a thermocouple placed
at the same position as the crystal. An arbitrary hemisphere of data was collected using
0.3° ω scans, and the data were integrated by the program SAINT.98 The final unit cell
parameters were determined by a least-squares refinement of the reflections with I >
2σ(I). Data analysis using Siemens XPREP99 and the successful solution and refinement
of the structure determined the space group. Empirical absorption corrections were
applied using the program SADABS.100 Equivalent reflections were averaged, and the
structure was solved by direct methods using the SHELXTL software package.101 All
non-hydrogen atoms were refined anisotropically.
4AgCl: X-ray quality crystals were grown from a pentane layered THF solution at
-25 °C. The final cycle of full-matrix least-squares refinement was based on 5605
observed reflections and 397 variable parameters and converged yielding final residuals:
R obs = 0.0266, R all = 0.0291, and GOF = 1.113.
69
Table 3.3 Crystallographic data for compound 4AgCl.
Compound
4AgCl
Formula
C 36 H 68 Ag 2 Cl 2 N 2 P 2
Formula weight
877.50
Space group
P2 1 /n
Crystal System
Monoclinic
Temperature (K)
140
a (Å)
16.0222(5)
b (Å)
13.0015(4)
c (Å)
20.2613(6)
α (°)
90.00
β (°)
101.220(1)
γ (°)
90.00
3
V (Å )
4140.0(2)
Z
4
3
Density calc (g/cm )
1.408
Diffractometer
Bruker APEX2
Radiation
Cu-K α (λ = 1.54178 Å)
Monochromator
Graphite
Detector
CCD detector
Scan type, width
ω, 0.3°
Scan speed (s)
10
Reflections measured
Hemisphere
2θ range (°)
7.82-117.86
Crystal dimensions (mm)
0.10 x 0.01 x 0.01
Reflections measured
24029
Unique reflections
5804
Observations (I > 2σ(I))
5605
R int
0.0297
Parameters
397
R obs , R w , R all
0.0266, 0.0740, 0.0291
GoF
1.113
70
References
(1)
Trost, B. M. Atom economy-a challenge for organic synthesis:
Homogeneous catalysis leads the way. Angewandte Chemie-International
Edition in English 1995, 34, 259-281.
(2)
Mueller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M.
Hydroamination: Direct addition of amines to alkenes and alkynes.
Chemical Reviews 2008, 108, 3795-3892.
(3)
Beck, J. F.; Schmidt, J. A. R. Hydroamination of 1,1-dimethylallene with
primary aryl amines under mild conditions: An atom-economical route to
N-(1,1-dimethyl-2-propenyl)-anilines. RSC Advances 2012, 2, 128-131.
(4)
Kinder, R. E.; Zhang, Z.; Widenhoefer, R. A. Intermolecular
hydroamination of allenes with N-unsubstituted carbamates catalyzed by a
gold(I) N-heterocyclic carbene complex. Organic Letters 2008, 10, 31573159.
(5)
Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-Catalyzed Hydroamination of
Alkynes and Allenes with Parent Hydrazine. Angewandte ChemieInternational Edition 2011, 50, 5560-5563.
(6)
Kojima, M.; Mikami, K. Enantioselective Intramolecular Hydroamination
of N-Alkenyl Ureas Catalyzed by tropos BIPHEP-Gold(I) Complexes with
Au-Au Interaction. Synlett 2012, 23, 57-61.
(7)
Yeh, M. C. P.; Pai, H. F.; Lin, Z. J.; Lee, B. R. Stereoselective synthesis of
hexahydroindoles and octahydrocyclohepta-[b]pyrroles via gold(I)catalyzed intramolecular 1,4-hydroamination of cyclic 1,3-dienes.
Tetrahedron 2009, 65, 4789-4794.
71
(8)
Hultzsch, K. C. Transition metal-catalyzed asymmetric hydroamination of
alkenes (AHA). Advanced Synthesis & Catalysis 2005, 347, 367-391.
(9)
Hultzsch, K. C. Catalytic asymmetric hydroamination of non-activated
olefins. Organic & Biomolecular Chemistry 2005, 3, 1819-1824.
(10)
Doye, S. Development of the Ti-catalyzed intermolecular hydroamination
of alkynes. Synlett 2004, 1653-1672.
(11)
Collet, F.; Lescot, C.; Dauban, P. Catalytic C-H amination: the
stereoselectivity issue. Chemical Society Reviews 2011, 40, 1926-1936.
(12)
Wang, Z. J.; Benitez, D.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D.
Mechanistic Study of Gold(I)-Catalyzed Intermolecular Hydroamination
of Allenes. Journal of the American Chemical Society 2010, 132, 1306413071.
(13)
Liu, Z.; Yamamichi, H.; Madrahimov, S. T.; Hartwig, J. F. Rhodium
Phosphine--Arene Intermediates in the Hydroamination of Alkenes.
Journal of the American Chemical Society 2011, 133, 2772-2782.
(14)
Yi, C. S.; Yun, S. Y. Ruthenium-catalyzed intermolecular coupling
reactions of arylamines with ethylene and 1,3-dienes: Mechanistic insight
on hydroamination vs ortho-C-H bond activation. Organic Letters 2005, 7,
2181-2183.
(15)
Sevov, C. S.; Zhou, J.; Hartwig, J. F. Iridium-Catalyzed Intermolecular
Hydroamination of Unactivated Aliphatic Alkenes with Amides and
Sulfonamides. Journal of the American Chemical Society 2012, 134,
11960-11963.
(16)
Walsh, P. J.; Baranger, A. M.; Bergman, R. G. Stochiometric and
Catalytic Hydroamination of Alkynes and Allene by Zirconium Bisamides
Cp 2 Zr(NHR) 2 . Journal of the American Chemical Society 1992, 114,
1708-1719.
(17)
Bytschkov, I.; Doye, S. Group-IV metal complexes as hydroamination
catalysts. European Journal of Organic Chemistry 2003, 935-946.
72
(18)
Ayinla, R. O.; Schafer, L. L. Intermolecular hydroamination of oxygensubstituted allenes. New routes for the synthesis of N,O-chelated
zirconium and titanium amido complexes. Dalton Transactions 2011, 40,
7769-7776.
(19)
Cooke, M. L.; Xu, K.; Breit, B. Enantioselective rhodium-catalyzed
synthesis of branched allylic amines by intermolecular hydroamination of
terminal allenes. Angewandte Chemie International edition in English
2012, 51, 10876-10879.
(20)
Duncan, A. N.; Widenhoefer, R. A. Gold(I)-Catalyzed Intermolecular
Hydroamination of Allenes with Arylamines. Synlett 2010, 419-422.
(21)
Zeng, X.; Soleilhavoup, M.; Bertrand, G. Gold-Catalyzed Intermolecular
Markovnikov Hydroamination of Allenes with Secondary Amines.
Organic Letters 2009, 11, 3166-3169.
(22)
Nishina,
N.;
Yamamoto,
Y.
Gold-catalyzed
intermolecular
hydroamination of allenes: First example of the use of an aliphatic amine
in hydroamination. Synlett 2007, 1767-1770.
(23)
Yuen, H. F.; Marks, T. J. Phenylene-Bridged Binuclear Organolanthanide
Complexes as Catalysts for Intramolecular and Intermolecular
Hydroamination. Organometallics 2009, 28, 2423-2440.
(24)
Li, Y. W.; Marks, T. J. Diverse mechanistic pathways and selectivities in
organo-f-element-catalyzed
hydroamination.
Intermolecular
organolanthanide-catalyzed alkyne and alkene hydroamination.
Organometallics 1996, 15, 3770-3772.
(25)
Koller, J.; Bergman, R. G. Aluminium-catalyzed intramolecular
hydroamination of aminoalkenes. Chemical Communications 2010, 46,
4577-4579.
(26)
Brinkmann, C.; Barrett, A. G. M.; Hill, M. S.; Procopiou, P. A. Heavier
Alkaline Earth Catalysts for the Intermolecular Hydroamination of
Vinylarenes, Dienes, and Alkynes. Journal of the American Chemical
Society 2012, 134, 2193-2207.
73
(27)
Dzhemilev, U.; Tolstikov, G.; Khusnutdinov, R. Hydroamination of
conjugated dienes catalyzed by transition metal complexes. Russian
Journal of Organic Chemistry 2009, 45, 957-987.
(28)
Severin, R.; Doye, S. The catalytic hydroamination of alkynes. Chemical
Society Reviews 2007, 36, 1407-1420.
(29)
Pan, S.; Endo, K.; Shibata, T. Ir(I)-Catalyzed Intermolecular Regio- and
Enantioselective Hydroamination of Alkenes with Heteroaromatic
Amines. Organic Letters 2012, 14, 780-783.
(30)
Giner, X.; Najera, C. Silver-Catalyzed Intermolecular Hydroamination of
Alkenes and 1,3-Dienes. Synlett 2009, 3211-3213.
(31)
Zhang, Z.; Lee, S. D.; Widenhoefer, R. A. Intermolecular Hydroamination
of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and
Chiral Gold(I) Complexes. Journal of the American Chemical Society
2009, 131, 5372-5373.
(32)
Zhou, J.; Hartwig, J. F. Intermolecular, catalytic asymmetric
hydroamination of bicyclic alkenes and dienes in high yield and
enantioselectivity. Journal of the American Chemical Society 2008, 130,
12220-12221.
(33)
Giner, X.; Najera, C. (Triphenyl phosphite)gold(I)-catalyzed
intermolecular hydroamination of alkenes and 1,3-dienes. Organic Letters
2008, 10, 2919-2922.
(34)
Brunet, J.-J.; Chu, N.-C.; Rodriguez-Zubiri, M. Platinum-catalyzed
intermolecular hydroamination of alkenes: Halide-anion-promoted
catalysis. European Journal of Inorganic Chemistry 2007, 4711-4722.
(35)
Rodriguez-Zubiri, M.; Anguille, S.; Brunet, J.-J. Intermolecular
hydroamination of non-activated alkenes catalyzed by Pt(II) or Pt(IV)-nBu 4 PX (X = Cl, Br, I) systems: Key effect of the halide anion. Journal of
Molecular Catalysis A: Chemical 2007, 271, 145-150.
(36)
Huang, J.-M.; Wong, C.-M.; Xu, F.-X.; Loh, T.-P. InBr 3 catalyzed
intermolecular hydroamination of unactivated alkenes. Tetrahedron
Letters 2007, 48, 3375-3377.
74
(37)
Taylor, J. G.; Whittall, N.; Hii, K. K. Copper-catalyzed intermolecular
hydroamination of alkenes. Organic Letters 2006, 8, 3561-3564.
(38)
Brunet, J. J.; Chu, N. C.; Diallo, O. Intermolecular highly regioselective
hydroamination of alkenes with ligandless Platinum(II) catalysts in ionic
solvents: Activation role of n-Bu 4 PBr. Organometallics 2005, 24, 31043110.
(39)
Wang, X.; Widenhoefer, R. A. Platinum-catalyzed intermolecular
hydroamination
of
unactivated
olefins
with
carboxamides.
Organometallics 2004, 23, 1649-1651.
(40)
Qian, H.; Widenhoefer, R. A. Platinum-catalyzed intermolecular
hydroamination of vinyl arenes with carboxamides. Organic Letters 2005,
7, 2635-2638.
(41)
Zhang, X.; Yang, B.; Li, G.; Shu, X.; Mungra, D. C.; Zhu, J. Highly
Regioselective AgNTf 2 -Catalyzed Intermolecular Hydroamination of
Alkynes with Anilines. Synlett 2012, 622-626.
(42)
Hild, F.; Dagorne, S. A Discrete N,O,N-Supported Gallium Amido
Complex for the Intermolecular Hydroamination of Terminal Alkynes.
Organometallics 2012, 31, 1189-1194.
(43)
Leyva-Perez, A.; Cabrero-Antonino, J. R.; Cantin, A.; Corma, A. Gold(I)
Catalyzes the Intermolecular Hydroamination of Alkynes with Imines and
Produces ,',N-Triarylbisenamines: Studies on Their Use As
Intermediates in Synthesis. Journal of Organic Chemistry 2010, 75, 77697780.
(44)
Leyva, A.; Corma, A. Reusable Gold(I) Catalysts with Unique
Regioselectivity for Intermolecular Hydroamination of Alkynes. Advanced
Synthesis & Catalysis 2009, 351, 2876-2886.
(45)
Dash, C.; Shaikh, M. M.; Butcher, R. J.; Ghosh, P. Highly Convenient
Regioselective Intermolecular Hydroamination of Alkynes Yielding
Ketimines Catalyzed by Gold(I) Complexes of 1,2,4-triazole Based Nheterocyclic Carbenes. Inorganic Chemistry 2010, 49, 4972-4983.
75
(46)
Cui, D.-M.; Zheng, J.-Z.; Yang, L.-Y.; Zhang, C. (PPh 3 )AuCl/AgOTfCatalyzed Intermolecular Hydroamination of Alkynes with Sulfonamides
To Form N-Sulfonyl Imines. Synlett 2010, 809-811.
(47)
Zeng, X.; Frey, G. D.; Kousar, S.; Bertrand, G. A Cationic Gold(I)
Complex as a General Catalyst for the Intermolecular Hydroamination of
Alkynes: Application to the One-Pot Synthesis of Allenes from Two
Alkynes and a Sacrificial Amine. Chemistry-a European Journal 2009,
15, 3056-3060.
(48)
Alex, K.; Tillack, A.; Schwarz, N.; Beller, M. General zinc-catalyzed
intermolecular hydroamination of terminal alkynes. Chemsuschem 2008,
1, 333-338.
(49)
Lai, R.-Y.; Surekha, K.; Hayashi, A.; Ozawa, F.; Liu, Y.-H.; Peng, S.-M.;
Liu, S.-T. Intra- and intermolecular hydroamination of alkynes catalyzed
by ortho-metalated iridium complexes. Organometallics 2007, 26, 10621068.
(50)
Brunet, J. J.; Chu, N. C.; Diallo, O.; Vincendeau, S. Platinum-catalyzed
intermolecular hydroamination of terminal alkynes. Journal of Molecular
Catalysis A: Chemical 2005, 240, 245-248.
(51)
Shimada, T.; Bajracharya, G. B.; Yamamoto, Y. Aquapalladium complex:
A stable and convenient catalyst for the intermolecular hydroamination of
alkynes. European Journal of Organic Chemistry 2004, 59-62.
(52)
Klein, D. P.; Ellern, A.; Angelici, R. J. New mechanism for the
intermolecular hydroamination of alkynes: Catalysis by dinuclear
ruthenium complexes with a rigid dicyclopentadienyl ligand.
Organometallics 2004, 23, 5662-5670.
(53)
Mizushima, E.; Hayashi, T.; Tanaka, M. Au(I)-catalyzed highly efficient
intermolecular hydroamination of alkynes. Organic Letters 2003, 5, 33493352.
(54)
Shimada, T.; Yamamoto, Y. Palladium-catalyzed intermolecular
hydroamination of alkynes: A dramatic rate-enhancement effect of oaminophenol. Journal of the American Chemical Society 2002, 124,
12670-12671.
76
(55)
Hartung, C. G.; Tillack, A.; Trauthwein, H.; Beller, M. A convenient
rhodium-catalyzed intermolecular hydroamination procedure for terminal
alkynes. Journal of Organic Chemistry 2001, 66, 6339-6343.
(56)
Tokunaga, M.; Eckert, M.; Wakatsuki, Y. Ruthenium-catalyzed
intermolecular hydroamination of terminal alkynes with anilines: A
practical synthesis of aromatic ketimines. Angewandte ChemieInternational Edition 1999, 38, 3222-3225.
(57)
Lavallo, V.; Frey, G. D.; Donnadieu, B.; Soleilhavoup, M.; Bertrand, G.
Homogeneous catalytic hydroamination of alkynes and allenes with
ammonia. Angewandte Chemie-International Edition 2008, 47, 52245228.
(58)
Nishina,
N.;
Yamamoto,
Y.
Gold-catalyzed
intermolecular
hydroamination of allenes with arylamines and resulting high chirality
transfer. Angewandte Chemie-International Edition 2006, 45, 3314-3317.
(59)
Patil, N. T.; Lutete, L. M.; Nishina, N.; Yamamoto, Y. Gold-catalyzed
intramolecular hydroamination of allenes: a case of chirality transfer.
Tetrahedron Letters 2006, 47, 4749-4751.
(60)
LaLonde, R. L.; Sherry, B. D.; Kang, E. J.; Toste, F. D. Gold(I)-catalyzed
enantioselective intramolecular hydroamination of allenes. Journal of the
American Chemical Society 2007, 129, 2452-2453.
(61)
Johns, A. M.; Liu, Z.; Hartwig, J. F. Primary tert- and sec-allylamines via
palladium-catalyzed hydroamination and allylic substitution with
hydrazine and hydroxylamine derivatives. Angewandte ChemieInternational Edition 2007, 46, 7259-7261.
(62)
Qiu, S.; Wei, Y.; Liu, G. Palladium-Catalyzed Intramolecular
Hydroamination of Allenes Coupled to Aerobic Alcohol Oxidation.
Chemistry-a European Journal 2009, 15, 2751-2754.
(63)
Ackermann, L.; Bergman, R. G.; Loy, R. N. Use of group 4
bis(sulfonamido) complexes in the intramolecular hydroamination of
alkynes and allenes. Journal of the American Chemical Society 2003, 125,
11956-11963.
77
(64)
Kanno, O.; Kuriyama, W.; Wang, Z. J.; Toste, F. D. Regio- and
Enantioselective Hydroamination of Dienes by Gold(I)/Menthol
Cooperative Catalysis. Angewandte Chemie-International Edition 2011,
50, 9919-9922.
(65)
Baeza, A.; Najera, C. Intramolecular Hydroamination of Conjugated
Dienes Catalyzed by Lewis and Bronsted Acids. Synlett 2011, 631-634.
(66)
Brouwer, C.; He, C. Efficient gold-catalyzed hydroamination of 1,3dienes. Angewandte Chemie-International Edition 2006, 45, 1744-1747.
(67)
Johns, A. M.; Utsunomiya, M.; Incarvito, C. D.; Hartwig, J. F. A highly
active palladium catalyst for intermolecular hydroamination. Factors that
control reactivity and additions of functionalized anilines to dienes and
vinylarenes. Journal of the American Chemical Society 2006, 128, 18281839.
(68)
Lober, O.; Kawatsura, M.; Hartwig, J. F. Palladium-catalyzed
hydroamination of 1,3-dienes: A colorimetric assay and enantioselective
additions. Journal of the American Chemical Society 2001, 123, 43664367.
(69)
Minami, T.; Okamoto, H.; Ikeda, S.; Tanaka, R.; Ozawa, F.; Yoshifuji, M.
(3-Allyl)palladium complexes bearing diphosphinidenecyclobutene
ligands: Highly active catalysts for the hydroamination of 1,3-dienes.
Angewandte Chemie-International Edition 2001, 40, 4501-4503.
(70)
Pawlas, J.; Nakao, Y.; Kawatsura, M.; Hartwig, J. F. A general nickelcatalyzed hydroamination of 1,3-dienes by alkylamines: Catalyst
selection, scope, and mechanism. Journal of the American Chemical
Society 2002, 124, 3669-3679.
(71)
Kuchenbeiser, G.; Shaffer, A. R.; Zingales, N. C.; Beck, J. F.; Schmidt, J.
A. R. Palladium(II) 3-iminophosphine (3IP) complexes: Active
precatalysts for the intermolecular hydroamination of 1,2-dienes (allenes)
and 1,3-dienes with aliphatic amines under mild conditions. Journal of
Organometallic Chemistry 2011, 696, 179-187.
78
(72)
Griller, D.; Kanabuskaminska, J. M.; Maccoll, A. Bond-Dissociation
Energies for Common Organic-Compounds. Theochem-Journal of
Molecular Structure 1988, 40, 125-131.
(73)
Benson, S. W. Bond Energies. Journal of Chemical Education 1965, 42,
502-515.
(74)
Johns, A. M.; Sakai, N.; Ridder, A.; Hartwig, J. F. Direct measurement of
the thermodynamics of vinylarene hydroamination. Journal of the
American Chemical Society 2006, 128, 9306-9307.
(75)
Li, H.; Du Lee, S.; Widenhoefer, R. A. Gold(I)-catalyzed enantioselective
intramolecular hydroamination of allenes with ureas. Journal of
Organometallic Chemistry 2011, 696, 316-320.
(76)
Tobisch, S. Intramolecular hydroamination/cyclisation of aminoallenes
mediated by a cationic zirconocene catalyst: a computational mechanistic
study. Dalton Transactions 2006, 4277-4285.
(77)
Jones, C. M.; Li, H.; Hickman, A. J.; Hughs, L. D.; Sobelman, S. J.;
Johnson, A. R. Sterically encumbered chiral amino alcohols for the
titanium catalyzed asymmetric alkylation of benzaldehyde. TetrahedronAsymmetry 2012, 23, 501-507.
(78)
Alcaide, B.; Almendros, P. Novel Cyclization Reactions of Aminoallenes.
Advanced Synthesis & Catalysis 2011, 353, 2561-2576.
(79)
Toups, K. L.; Widenhoefer, R. A. Platinum(II)-catalyzed intermolecular
hydroamination of monosubstituted allenes with secondary alkylamines.
Chemical Communications 2010, 46, 1712-1714.
(80)
Butler, K. L.; Tragni, M.; Widenhoefer, R. A. Gold(I)-Catalyzed
Stereoconvergent, Intermolecular Enantioselective Hydroamination of
Allenes. Angewandte Chemie-International Edition 2012, 51, 5175-5178.
(81)
Ayinla, R. O.; Schafer, L. L. Bis(amidate) titanium precatalyst for the
intermolecular hydroamination of allenes. Inorganica Chimica Acta 2006,
359, 3097-3102.
79
(82)
Lingaiah, N.; Babu, N. S.; Reddy, K. M.; Prasad, P. S. S.; Suryanarayana,
I. An efficient reusable silver-exchanged tungstophosphoric acid
heterogeneous catalyst for solvent-free intermolecular hydroamination of
alkynes. Chemical Communications 2007, 278-279.
(83)
McBee, J. L.; Bell, A. T.; Tilley, T. D. Mechanistic Studies of the
Hydroamination of Norbornene with Electrophilic Platinum Complexes:
The Role of Proton Transfer. Journal of the American Chemical Society
2008, 130, 16562-16571.
(84)
Brunet, J. J.; Chu, N. C.; Diallo, O.; Mothes, E. Catalytic intermolecular
hydroamination of alkenes - Beneficial effect of ionic solvents. Journal of
Molecular Catalysis A: Chemical 2003, 198, 107-110.
(85)
Beller, M.; Thiel, O. R.; Trauthwein, H.; Hartung, C. G. AntiMarkovnikov functionalizations of unsaturated compounds, Part 7 Amination of aromatic olefins with anilines: A new domino synthesis of
quinolines. Chemistry-a European Journal 2000, 6, 2513-2522.
(86)
Dub, P. A.; Rodriguez-Zubiri, M.; Baudequin, C.; Poli, R.
Hydroamination of ethylene by aniline: catalysis in water. Green
Chemistry 2010, 12, 1392-1396.
(87)
Brunet, J. J.; Cadena, M.; Chu, N. C.; Diallo, O.; Jacob, K.; Mothes, E.
The first platinum-catalyzed hydroamination of ethylene. Organometallics
2004, 23, 1264-1268.
(88)
Shaffer, A. R.; Schmidt, J. A. R. Palladium(II) 3-iminophosphine
complexes as intermolecular hydroamination catalysts for the formation of
imines and enamines. Organometallics 2008, 27, 1259-1266.
(89)
Scheideman, M.; Wang, G. Q.; Vedejs, E. Amine-directed hydroboration:
Scope and limitations. Journal of the American Chemical Society 2008,
130, 8669-8676.
(90)
Worthy, A. D.; Joe, C. L.; Lightburn, T. E.; Tan, K. L. Application of a
Chiral
Scaffolding
Ligand
in
Catalytic
Enantioselective
Hydroformylation. Journal of the American Chemical Society 2010, 132,
14757-14759.
80
(91)
Grubbs, R. H. Olefin metathesis. Tetrahedron 2004, 60, 7117-7140.
(92)
Johannsen, M.; Jorgensen, K. A. Allylic amination. Chemical Reviews
1998, 98, 1689-1708.
(93)
Paquette, L. A. Johnson, B. A.; Hinga, F. M. 2-chloro-1-formyl-1cyclohexene. Col. Org. Syn. 1973, 5, 18-20.
(94)
Frew, J. J. R.; Damian, K.; Van Rensburg, H.; Slawin, A. M. Z.; Tooze, R.
P.; Clarke, M. L. Palladium(II) Complexes of New Bulky Bidentate
Phosphanes: Active and Highly Regioselective Catalysts for the
Hydroxycarbonylation of Styrene. Chemistry-a European Journal 2009,
15, 10504-10513.
(95)
Steffen, W. L.; Palenik, G. J. Crystal and Molecular Structures of
Dichloro[bis(diphenylphosphino)methane]palladium(II),
Dichloro[bis(diphenylphosphino) ethane]palladium(II), and Dichloro[1,3bis(diphenylphosphino)propane]palladium(II). Inorganic Chemistry 1976,
15, 2432-2439.
(96)
Diamond: Crystal and Molecular Structure Visualization and Exploration
Program 3.2 ed.; Crystal Impact: Bonn, Germany, 2012.
(97)
SMART: Area-Detector Software Package; 5.625 ed.; Bruker AXS, Inc.:
Madison, WI, 1997-2001.
(98)
SAINT: SAX Area-Detector Integration Program; 6.22 ed.; Bruker AXS,
Inc.: Madison, WI, 1997-2001.
(99)
XPREP: Reciprocal Space Exploration Program; 6.12 ed.; Bruker AXS,
Inc.: Madison, WI, 2001.
(100) SADABS: Bruker/Siemens Area Detector Absorption Program; 2.03 ed.;
Bruker AXS, Inc.: Madison, WI, 2001.
(101) SHELXTL-97, Structure Solution Program; 6.10 ed.; Bruker AXS, Inc.:
Madison, WI, 2000.
81
(102) Zingales, N. C.; Shaffer, A. R.; Schmidt, J. A. R. Investigation of Steric
and Electronic Features of 3-Iminophosphine-Based Palladium Catalysts
for Intermolecular Hydroamination. Organometallics 2013, 32, submitted.
(103) Bowmaker, G. A.; Effendy; Harvey, P. J.; Healy, P. C.; Skelton, B. W.;
White, A. H. Spectroscopic and structural studies on 1:1 adducts of
silver(I) salts with tricyclohexylphosphine. Journal of the Chemical
Society-Dalton Transactions 1996, 2459-2465.
(104) Tolman, C. A. Steric Effects of Phosphorus Ligands in Organometallic
Chemistry and Homogeneous Catalysis. Chemical Reviews 1977, 77, 313348.
(105) Shaffer, A. R.; Schmidt, J. A. R. A Versatile Methodology for the
Synthesis of alpha,beta-Unsaturated 3-Iminophosphines. Chemistry-a
European Journal 2009, 15, 2662-2673.
82