Climbing in the tree of life
... When Human Genomes were first sequenced it was shown that the average number of single nucleotide polymorphisms between any two people was around 0.1% of the entire genome. With only a few genomes sequenced this meant that there were a few million SNPs discovered ...
... When Human Genomes were first sequenced it was shown that the average number of single nucleotide polymorphisms between any two people was around 0.1% of the entire genome. With only a few genomes sequenced this meant that there were a few million SNPs discovered ...

Evolutionary Representations of Biological History
... Nowadays molecular data are mostly used to reconstruct species trees. Molecular evolution or phylogenetics is the branch of biology that reconstructs evolutionary histories of species and genes by using molecular data, like DNA sequences. Not all DNA sequences can be used to reconstruct trees, but o ...
... Nowadays molecular data are mostly used to reconstruct species trees. Molecular evolution or phylogenetics is the branch of biology that reconstructs evolutionary histories of species and genes by using molecular data, like DNA sequences. Not all DNA sequences can be used to reconstruct trees, but o ...

You can position your opening statement here, either in
... • Single Nucleotide Polymorphisms (SNPs) are DNA sequence variations that occur when a single nucleotide (A,T,C,or G) is changed, which occur approximately once every 100 to 300 bases • The resulting different forms of the same gene are called Alleles. People can have two identical or two different ...
... • Single Nucleotide Polymorphisms (SNPs) are DNA sequence variations that occur when a single nucleotide (A,T,C,or G) is changed, which occur approximately once every 100 to 300 bases • The resulting different forms of the same gene are called Alleles. People can have two identical or two different ...

Microevolutionary processes in the stygobitic genus Typhlocirolana
... Morocco with several species. More populations have recently been found in Morocco, in some southern regions around Agadir, in High Atlas valleys near Marrakech and in the northeastern part of the country close to Oujda. The populations of these zones are not yet described and are the subject of thi ...
... Morocco with several species. More populations have recently been found in Morocco, in some southern regions around Agadir, in High Atlas valleys near Marrakech and in the northeastern part of the country close to Oujda. The populations of these zones are not yet described and are the subject of thi ...

doc - BeanBeetles.org
... Use multiple trees to model the most likely evolutionary relationship between taxa. ...
... Use multiple trees to model the most likely evolutionary relationship between taxa. ...

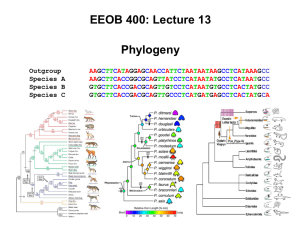
characters
... Some horned lizards squirt blood from their eyes when attacked by canids How many times has blood-squirting evolved? This phylogeny suggests a single evolutioary gain and a single loss of blood squirting ...
... Some horned lizards squirt blood from their eyes when attacked by canids How many times has blood-squirting evolved? This phylogeny suggests a single evolutioary gain and a single loss of blood squirting ...

Cladogram Activity
... LO 1.17 The student is able to pose scientific questions about a group of organisms whose relatedness is described by a phylogenetic tree or cladogram in order to (1) identify shared characteristics, (2) make inferences about the evolutionary history of the group, and (3) identify character data tha ...
... LO 1.17 The student is able to pose scientific questions about a group of organisms whose relatedness is described by a phylogenetic tree or cladogram in order to (1) identify shared characteristics, (2) make inferences about the evolutionary history of the group, and (3) identify character data tha ...

Comparing data sets It is possible to collect multiple different data
... doesn’t worry so much about the topology of the optimal or plausible trees but rather focuses on the length of the MP trees for each partition. Recall (PTP and skewness tests, page xx) that as we decrease the phylogenetic signal within a data set we tend to increase the length of the optimal tree be ...
... doesn’t worry so much about the topology of the optimal or plausible trees but rather focuses on the length of the MP trees for each partition. Recall (PTP and skewness tests, page xx) that as we decrease the phylogenetic signal within a data set we tend to increase the length of the optimal tree be ...

Molecular evidence for the origin of birds
... cytochrome c, insulin, and tRNAVal,the small number of variable (and parsimony) sites cannot be expected to give reliable results in a phylogenetic analysis, as indicated by the alternative relationships obtained when additional taxa are included. Until now, sequences from P-hemoglobin have provided ...
... cytochrome c, insulin, and tRNAVal,the small number of variable (and parsimony) sites cannot be expected to give reliable results in a phylogenetic analysis, as indicated by the alternative relationships obtained when additional taxa are included. Until now, sequences from P-hemoglobin have provided ...

ppt - Chair of Computational Biology
... supported only a few alternative trees, (2) most genes could have strongly supported one phylogeny and a few genes strongly supported only a small number of alternatives, (3) there could have been some combinations of these scenarios so that each branch among alternative phylogenies had either weak ...
... supported only a few alternative trees, (2) most genes could have strongly supported one phylogeny and a few genes strongly supported only a small number of alternatives, (3) there could have been some combinations of these scenarios so that each branch among alternative phylogenies had either weak ...

Chapter 25.
... An unexpected family tree. What are the evolutionary relationships among a human, a mushroom, and a tulip? Molecular systematics has revealed that— ...
... An unexpected family tree. What are the evolutionary relationships among a human, a mushroom, and a tulip? Molecular systematics has revealed that— ...
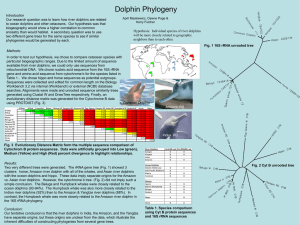
dolphin1
... Our research question was to learn how river dolphins are related to ocean dolphins and other cetaceans. Our hypothesis was that biogeography would show a higher correlation to common ancestry than would habitat. A secondary question was to use two different gene trees for the same species to see if ...
... Our research question was to learn how river dolphins are related to ocean dolphins and other cetaceans. Our hypothesis was that biogeography would show a higher correlation to common ancestry than would habitat. A secondary question was to use two different gene trees for the same species to see if ...

lecture 9
... ¾ COG specific phylogenetic profiles can be used to predict functional association of among COGs, and the living styles of ...
... ¾ COG specific phylogenetic profiles can be used to predict functional association of among COGs, and the living styles of ...


manual
... -l LSCALE, --length scale=LSCALE Sequence lengths scaling for alignments. Each alignment in each simulation can be scaled with the same number. -v THETA, --theta=THETA θ parameter for generating gene trees from the species trees before generating alignments along the species tree. For the definition ...
... -l LSCALE, --length scale=LSCALE Sequence lengths scaling for alignments. Each alignment in each simulation can be scaled with the same number. -v THETA, --theta=THETA θ parameter for generating gene trees from the species trees before generating alignments along the species tree. For the definition ...

Species Tree and Most Likely Gene Tree
... are short, frequently there isn’t enough information about that specific branching because very few mutations happen along it. In those cases, anomalous gene trees aren’t even an issue. So in actual sequence analysis, anomalous gene trees may only come up when these short internal branches have high ...
... are short, frequently there isn’t enough information about that specific branching because very few mutations happen along it. In those cases, anomalous gene trees aren’t even an issue. So in actual sequence analysis, anomalous gene trees may only come up when these short internal branches have high ...

Document
... Problem: Long Branch Attraction (LBA) • Particular problem associated with parsimony methods • Rapidly evolving taxa are placed together in a tree regardless of their true position • Partly due to assumption in parsimony that all lineages evolve at the same rate • This means that also UPGMA suffers ...
... Problem: Long Branch Attraction (LBA) • Particular problem associated with parsimony methods • Rapidly evolving taxa are placed together in a tree regardless of their true position • Partly due to assumption in parsimony that all lineages evolve at the same rate • This means that also UPGMA suffers ...

Supplemental Data
... Figure S3. RT-PCR analysis of the irx9 and irx9-L1 alleles RT-PCR was performed on RNA prepared from irx9-L1, irx9 and irx9-L1 irx9 plants using primers specific for IRX9-L and IRX9. Primers for the 18S rRNA were used as a control. ...
... Figure S3. RT-PCR analysis of the irx9 and irx9-L1 alleles RT-PCR was performed on RNA prepared from irx9-L1, irx9 and irx9-L1 irx9 plants using primers specific for IRX9-L and IRX9. Primers for the 18S rRNA were used as a control. ...

my_phylogeny1
... with the fewest evolutionary changes for all sequences to derive from a common ancestor. Phylip • Slower than distance methods. • Assumes molecular clock • Maximum Likelihood : Looks for the tree with the maximum likelihood: the most probable tree. • this is the slowest method of all but seems to gi ...
... with the fewest evolutionary changes for all sequences to derive from a common ancestor. Phylip • Slower than distance methods. • Assumes molecular clock • Maximum Likelihood : Looks for the tree with the maximum likelihood: the most probable tree. • this is the slowest method of all but seems to gi ...
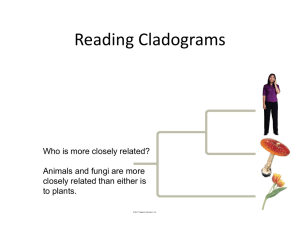
Reading Cladograms
... • Two species are closely related if their most recent common ancestor lived closer to the present • More distantly related if their most recent common ancestor lived in the distant past ...
... • Two species are closely related if their most recent common ancestor lived closer to the present • More distantly related if their most recent common ancestor lived in the distant past ...

Mine Microarray Gene Expression Data, Predict Cancers
... • Single gene (zyxin), single branch tree 38/38 correct on training cases 31/34 correct on test cases, 3 errors X*5735_at <=(8+1)38: ALL • Tree size up to 3 genes 1 decision tree with 1 error 7 decision trees with 2 errors 7 decision trees with 3 errors ...
... • Single gene (zyxin), single branch tree 38/38 correct on training cases 31/34 correct on test cases, 3 errors X*5735_at <=(8+1)38: ALL • Tree size up to 3 genes 1 decision tree with 1 error 7 decision trees with 2 errors 7 decision trees with 3 errors ...

Phylogenetic Relationships Among Ascomycetes: Evidence from an
... pCR2.1 plasmid vector (Invitrogen Inc., Carlsbad, Calif.). A number of clones for each RPB2 fragment were subjected to automatic sequencing (ABI PRISM Dye Terminator Cycle Sequencing and ABI PRISM Sequencer model 377, Perkin-Elmer). Fungal-specific primers were then designed based on the RPB2 sequen ...
... pCR2.1 plasmid vector (Invitrogen Inc., Carlsbad, Calif.). A number of clones for each RPB2 fragment were subjected to automatic sequencing (ABI PRISM Dye Terminator Cycle Sequencing and ABI PRISM Sequencer model 377, Perkin-Elmer). Fungal-specific primers were then designed based on the RPB2 sequen ...
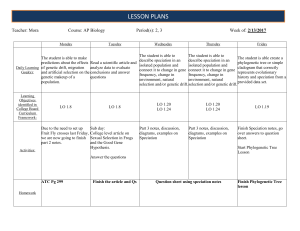
lesson Plans - Lemon Bay High School
... Due to the need to set up Fruit Fly crosses last Friday, we are now going to finish part 2 notes. Activities: ...
... Due to the need to set up Fruit Fly crosses last Friday, we are now going to finish part 2 notes. Activities: ...

15.16 Shared characters are used to construct phylogenetic trees
... – can be calibrated in real time by graphing the number of nucleotide differences against the dates of evolutionary branch points known from the fossil record, – are used to estimate dates of divergences without a good fossil record, and – have been used to date the origin of HIV infection in ...
... – can be calibrated in real time by graphing the number of nucleotide differences against the dates of evolutionary branch points known from the fossil record, – are used to estimate dates of divergences without a good fossil record, and – have been used to date the origin of HIV infection in ...