Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Synthetic biology wikipedia , lookup
History of genetic engineering wikipedia , lookup
Designer baby wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Microevolution wikipedia , lookup
Cancer epigenetics wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Genome (book) wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Mir-92 microRNA precursor family wikipedia , lookup
Secreted frizzled-related protein 1 wikipedia , lookup
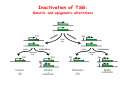
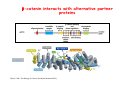
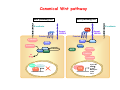
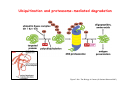
Robert A. Weinberg The Biology of Cancer First Edition Chapter 7: Tumor Suppressor Genes Copyright © Garland Science 2007 張久瑗 中央研究院 生物醫學研究所 副研究員 1. Cancer Genomics 2. Tumor and Tel. (02)2789-9046 Molecular Biology Fax. (02)2785-8594 [email protected] Outlines: -Cell fusion: indicating that the cancer phenotype is recessive -The retinoblastoma tumor -Familial vs. sporadic forms -Knudson’s two-hit theory -Inactivation of TSGs -LOH -mutations -promoter hypermethylation -TSGs and proteins function in diverse ways -pRB (Chap. 8) and cell cycle control -p53 (Chap. 9) and apoptosis -NF1 protein acts as a negative regulator of Ras signaling -Apc facilitates egress of cells from colonic crypts -Von Hippel-Lindau disease: pVHL modulates the hypoxic response Hallmarks of Cancer The Hallmarks of Cancer Cell (2000) 100:57 Rules for Making Human Tumor Cells N Engl J Med (2002) 14:1593 Normal vs. Transformed Cells Normal 3T3 cells: Elongated Aligned and packed in an ordered fashion RSV-transformed Cells: more rounded and covered by hair-like processes Lost side-by-side organization Cell fusion Fusion of mouse 3T3 cells with monkey kidney cells (PEG or inactivated Sendai virus) heterokaryon (cultured under selection) (a polykaryon containing 9 nuclei) Figure 7.1 The Biology of Cancer (© Garland Science 2007) Identification of recessive cancer-inducing alleles (tumor suppressor genes) (non-tumorigenic) (from virus-induced tumors) (tumorigenic) (fom non-virus-induced human tumors, or from chemically induced rodent tumord) If the cancer cell had originally arisen without the involvement of a tumor virus, then its malignant phenotype was recessive when this cell was fused with a normal cell. Figure 7.3 The Biology of Cancer (© Garland Science 2007) Pediatric retinoblastoma tumor Sporadic form -unilateral and unifocal -once the tumor is eliminated, no further risk Familiar form -bilateral and often multi-focal -curing the eye tumor does not protect the children from a greatly increased risk to bone cancers and other cancers Pedigree of a kindred afflicted with Rb (Affected individual: green-filled circles or squares) -Conforms somatic dominant transmission pattern Figure 7.4The Biology of Cancer (© Garland Science 2007) Kinetics of Rb: unilateral vs. bilateral Two-hit hypothesis: Alfred Knudson concluded in 1971 that the rate of appearance of familial tumors was consistent with a single random event, while the sporadic tumors behaved as if two random events were required for their formation. Figure 7.6 The Biology of Cancer (© Garland Science 2007) Dynamics of retinoblastoma formation Figure 7.7 The Biology of Cancer (© Garland Science 2007) Germ-line vs. Somatic Mutations -bilateral -multi-focal -unilateral -uni-focal Knudson proposed that two “hits”, or mutagenic events, were necessary for retinoblastoma development in all cases. Incidence of developing non-retinal tumors of Rb patients -Among the 1601 Rb patients diagnosed between 1914-1984, those cured of bilateral tumors have a dramatically higher risk of developing second tumors Figure 7.4The Biology of Cancer (© Garland Science 2007) Knudson’s Two-Hit Hypothesis NORMAL HEALTHY INDIVIDUAL HEREDITARY RETINOBLASTOMA inherited mutant Rb1 gene occasional cell inactivates one of its two good Rb1 genes occasional cell inactivates its only good Rb1 gene copy NONHEREDITARY RETINOBLASTOMA occasional cell inactivates one of its two good Rb1 genes excessive cell proliferation leading to retinoblastoma excessive cell proliferation leading to retinoblastoma Mitotic recombination: a possible mechanism to eliminate the wild-type copy of Rb gene? The probability of inactivating a single gene copy by mutation is on the order of 10-6 per cell generation, the probability of silencing both copies is on the order of 10-12 per cell generation. It seems highly unlikely that both copies of the Rb gene could be eliminated through two recessive mutational event in the relatively small target cell populations in the developing retina (about 106 cells). Figure 7.8 The Biology of Cancer (© Garland Science 2007) Mutant Rb genes are both dominant and recessive. An individual who inherits a mutant, defective allele of Rb is almost certain to develop retinoblastoma at some point in childhood. However, a cell carrying a mutant and a wild-type Rb gene copy would behave normally. The mutant Rb allele acts dominantly at the organismic level and recessively at cellular level. Gene conversion: another possible mechanism to eliminate the wild-type copy of Rb gene? Figure 7.9 The Biology of Cancer (© Garland Science 2007) Chromosomal localization of Rb locus, 13q14 Figure 7.10 The Biology of Cancer (© Garland Science 2007) Loss of heterozygosity: esterase D locus serves as a surrogate marker for RB Zymographic analysis of esterase D isoforms Figure 7.11 The Biology of Cancer (© Garland Science 2007) Mutations of Rb gene Inactivation of Rb gene is achieved by the deletion of the wild-type allele accompanied with the mutations in the second allele. Figure 7.12 The Biology of Cancer (© Garland Science 2007) Genetic Markers (To qualify as a genetic marker, a locus must be polymorphic.) RFLP (Restriction Fragment Length Polymorphisms): -single-base or insertion/deletion polymorphisms lying within restriction enzyme recognition sites -detected as variations in the lengths of restriction fragments VNTR (Variable Number of Tandem Repeats) -VNTRs of a common motif of 16 nucleotides or greater Microsatellite polymorphisms (also called STRP, Short Tandem Repeat Polymorphisms) -Short tandem repeat motifs, term “microsatellites“ -arise from variation in the number of di-, tri-, or a larger number of nucleotides. -have a large number of alleles (and are thus highly polymorphic) -Normally occur once every 30 kb. -More than 8900 microsatellite markers that span the entire human genome have been identified, characterized, and mapped. SNP (Single Nucleotide Polymorphisms) -polymorphisms corresponding to differences at a single nucleotide position (e.g., substitutions, deletions, and insertions) -biallelic polymorphisms, generally less informative than STRPs.) -In human genome, an SNP with a hetrozygosity greater than 30% occurs, on average, approximately every 1.3 kb. -powerful tools for searching for complex disease genes by association study or linkage disequilibrium analysis Restriction fragment length polymorphism (RFLP) Figure 7.13 The Biology of Cancer (© Garland Science 2007) Loss of Heterozygosity: Identifying Lost Tumor Suppressor Genes Single nucleatide polymorphism (SNP) Allele-specific primer extension reaction Figure 7.15 The Biology of Cancer (© Garland Science 2007) LOH of chromosomal arms in CRC: High frequency of LOH allows the detection of putative TSG Figure 7.14 The Biology of Cancer (© Garland Science 2007) -These genes specify a diverse array of proteins that operate in many different intracellular sites to reduce the risk of cancer. -An anti-cancer function is the only property that is shared by these otherwise unrelated genes. -Many familial cancers can be explained by inheritance of mutant TSGs. -Inheritance of defective copies of TSGs creats an enormous risk of contracting one or another specific type of cancer. In some cases, mutant germline alleles of these genes lead to susceptibility to multiple cancer types. Table 7.1 The Biology of Cancer (© Garland Science 2007) Cancer Formation • Clonal Expansion -Tumor formation is driven by a multi-step process of genetic alterations. -Each event confers growth advantage to the expanded cell population. -Genetic alterations promote clonal evolution include activation of protooncogenes, inactivation of tumor suppressor genes, and de-regulation of cell cycle regulators, etc. • Mutator Phenotype -Pre-existing mutations in genes involved in the maintenance of genomic integrity drives cancer formation. -Increasing genetic instability is observed in the process of tumor progression. -Events contributing to mutator phenotype include errors in DNA replication, deficits in DNA repair, disregulation of checkpoint control, and abnormalities in chromosomal segregation, etc. Gatekeeper vs. Caretaker Gatekeeper are TSGs that directly control the biology of cells by affecting proliferation, differentiation, and apoptosis, and therefore. The name of gatekeeper indicate their role in allowing or disallowing cells to progress through cell cycles of growth and division. Inactivation of these genes is rate-limiting for the initiation of tumor formation, and both copies must be altered for tumor development. Caretaker are DNA maintenance genes that affect cell biology indirectly by controlling the rate at which cells accumulate mutant genes. The name of Caretaker reflects their role in the meintenance of cellular genomes. Inactivation of TSG: Genetic and epigenetic alterations Hypermethylation of RASSF1A promoter Figure 7.17 The Biology of Cancer (© Garland Science 2007) Table 7.2 The Biology of Cancer (© Garland Science 2007) Transcription silencing of TSGs in tumors Figure 7.19 The Biology of Cancer (© Garland Science 2007) NF1: a negative regulator of Ras signaling Figure 7.21 The Biology of Cancer (© Garland Science 2007) Familial adenomatous polyposis (FAP) the wall of colon from a FAP patient polyps the wall of a normal colon Adenomatous growths, polys in colon -Pedunculated polys are shown as stalk-like growths. Sessil polys appearing as flat thickenings of colonic wall are not shown. Figure 7.23 The Biology of Cancer (© Garland Science 2007) Disease Gene Localization Genetic mapping using large FAP kindreds Figure 7.23 The Biology of Cancer (© Garland Science 2007) Linkage analysis: association of phenotype to genotype the usage of polymorphic markers APC (adenomatosis polyposis coli) at chromosome 5q21 Figure 7.25a The Biology of Cancer (© Garland Science 2007) β-catenin interacts with alternative partner proteins APC β-catenin Figure 7.25b The Biology of Cancer (© Garland Science 2007) Canonical Wnt pathway Wnt pathway off Wnt pathway on E-cadherin E-cadherin Wnt Arrow/ LRP5/6 Arrow/ LRP5/6 Frizzled Frizzled β-catenin Dsh Axin Dsh β-catenin GSK3 Axin conductin GSK3 APC β-catenin APC β-catenin β-catenin HDAC Groucho TCF TCF Cyclin D1 PPARδ C-myc Matrilysin CD44 Tcf-1 Role of β-catenin/TCF pathway IHC of Ki67 enterocytes lumen of intestine mesenchymal core bottom of crypts TCF4+/- Figure 7.24a The Biology of Cancer (© Garland Science 2007) Role of β-catenin/TCF pathway IHC of β-catenin in Min mouse DAPI stains nuclei. Figure 7.24-25 The Biology of Cancer (© Garland Science 2007) IHC of Ki67 in TCF-mice Histopathology of colorectal cancer Nat Rev Cancer 1: 55 (2001) Mutation of APC/β-Catenin pathway in human colon tumors Mitotic spindle in relation to APC and CIN APC accumulates at the kinetochore through interaction with the microtubule-associated protein EB, where it may facilitate the binding of spindle microtubules to kinetochore. Interaction of kinetochores and spindle microtubule is disrupted, leading to chromosomal instability. Von Hippel-Lindau (VHL): LOH of 3q25 Nat Rev Cancer 2: 673 (2002) Ubiquitination and proteasome-mediated degradation Ubi Figure 7.26a The Biology of Cancer (© Garland Science 2007) Regulation of HIF-1 by pVHL angiogenesis pVHL is a component of an E3 ubiquitin ligase that targets the a subunits of HIF (hypoxia-inducible factor) transcription factor for destruction in the presence of oxygen. Figure 7.28a The Biology of Cancer (© Garland Science 2007) Table 7.3 The Biology of Cancer (© Garland Science 2007)