Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Antimicrobial peptides wikipedia , lookup
Human leukocyte antigen wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
Innate immune system wikipedia , lookup
Immune system wikipedia , lookup
Gluten immunochemistry wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Immunocontraception wikipedia , lookup
Adaptive immune system wikipedia , lookup
Duffy antigen system wikipedia , lookup
DNA vaccination wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
Major histocompatibility complex wikipedia , lookup
Monoclonal antibody wikipedia , lookup
The Immune Response
The humoral response
involves interaction of B
cells with antigen (Ag) and
their differentiation into
antibody-secreting plasma
cells. The secreted antibody
(Ab) binds to the antigen
and facilitates its clearance
from the body.
The cell-mediated
responses involve various
subpopulations of T cells
that recognize antigen
presented on self-cells.
Helper T cells respond to
antigen by producing
cytokines. Cytotoxic T cells
respond to antigen by
developing into cytotoxic T
lymphocytes (CTLs), which
mediate killing of altered
self-cells (e.g., virusinfected cells).
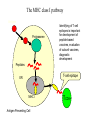
The MHC class I pathway
Antigen
Proteasome
Identifying of T-cell
epitopes is important
for development of
peptide-based
vaccines, evaluation
of subunit vaccines,
diagnostic
development
Peptides
T-cell epitope
ER
MHC I
Antigen Presenting Cell
TCD8+
The immunoglobulin fold
Common Structures - Both the antibodies
of the humoral response and the molecules
involved in the cellular response (antibody,
TCR, most CD [cell surface molecules
expressed on various cell types in the
immune system]) contain elements of
common structure.
The domains in these molecules are built
on a common motif, called the
immunoglobulin fold, in which two antiparallel sheets lie face to face. This
structure probably represents the primitive
structural element in the evolution of the
immune response. The immunoglobulin
fold is also found in a number of other
proteins.
Complex Of A Human TCR, Influenza HA Antigen
Peptide (PKYVKQNTLKLAT) and MHC Class II
Xenoreactive Complex AHIII 12.2 TCR bound
to P1049 (ALWGFFPVLS) /HLA-A2.1
Epitope, or antigenic determinant, is
defined as the site of an antigen
T-Cell
recognized by immune response
Receptor
molecules (antibodies, MHC, TCR)
T cell epitope – a short linear peptide or
other chemical entity (native or
denatured antigen) that binds MHC
V
V I binds 8-10 ac peptides; class II
(class
binds 11-25 ac peptides) and may be
recognized by T-cell receptor (TCR).
MHC
class II
T cell recognition of antigen involves
tertiary complex “antigen-TCR-MHC”.
MHC
class II
1fyt
T-Cell
Receptor
V
V
MHC
class I
-2-Microglobulin
1lp9
Igg2A Intact Mouse Antibody - Mab231 (PDB ID 1igt)
Fab fragment
VL
VH
CL
CH
Fv fragment
Light chain
Fc fragment
Heavy chain
B cell epitope – a site on
the surface of the
antigen structure that
binds antibody
molecule.
Protein antigens usually
contain both sequential (or
continues, they could work as
epitopes even when a protein
is denatured) and
nonsequential (discontinues
or conformational) epitopes.
B cell recognition of antigen
involves binary complex
“native antigen-membrane
immunoglobulin”.
Different antibody recognize
different epitopes.
Most of the surface of a
globular protein is potentially
antigenic.
Sperm whale myoglobin (1vxg) contains
five sequential epitopes (red, green,
magenta, blue, orange) and two
conformational epitopes (yellow, pink).
HIV-1 envelope protein
gp120 core complexed
with CD4 and a
neutralizing human
antibody 17b
HIV-1 envelope
protein gp120 (core
fragment)
The entry of HIV into cells
requires the sequential
interaction of the viral exterior
envelope glycoprotein, gp120,
with the CD4 glycoprotein and
CD4 (N-terminal
a chemokine receptor on the
two domain
cell surface. These
interactions initiate a fusion of fragment)
the viral and cellular
membranes. Although gp120
can elicit virus-neutralizing
antibodies, HIV eludes the
Antibody 17b
immune system.
(Fab fragment)
17b epitope is comprised of
four discontinuous -strands.
17b epitope
PDB: 1gc1
B cells and T cells recognize different epitopes
of the same protein antigen
T cell epitope
B cell epitope
Denatured antigen
Native or denatured (rare) antigen
Linear peptide 8-30 ac
Sequential or conformational
Internal (often)
Accessible, hydrophilic, mobile,
usually on the surface or could be
exposed as a result of
physicochemical change
Binding to T cell receptor:
Kd 10-5 – 10-7 M (low affinity)
Binding to antibody:
Slow on-rate, slow off-rate
Kd 10-7 – 10-11 M (high affinity)
(once bound, peptide may stay
associated for hours to many days)
Rapid on-rate, variable off-rate
Types of protein-protein interactions (PPI)
Non-obligate PPI
Obligate PPI
usually
permanent
the protomers are
not found as stable
structures on their
own in vivo
Permanent
Transient
(most enzyme-inhibitor
complexes)
Weak
dissociation constant
Kd = [A] [B] / [AB]
(electron
transport
complexes)
10-7 ÷ 10-13 M
Kd mM-M
Intermediate
Non-obligate transient
homodimer, Sperm lysin
(interaction is broken and
formed continuously)
(antibody-antigen, TCR-MHC-peptide,
signal transduction PPI), Kd M-nM
Strong
Obligate
heterodimer
Human cathepsin D
Non-obligate
permanent
heterodimer
(require a molecular
trigger to shift the
oligomeric
equilibrium)
Kd nM-fM
Thrombin and rodniin Bovine G protein dissociates into G and G subunits
inhibitor
upon GTP, but forms a stable trimer upon GDP
B cell (magenta, orange) and T cell epitopes (blue,
green, red) of hen egg-white lysozyme
PDB: 1dpx
Immune Epitope Database and Analysis Resource
IEDB, the newly developed
public database by the LIAI
together with the SAIC,
UCSD, and Denmark
University and sponsored
by the NIH, maintains
experimental data on
immune epitopes (the sites
on foreign molecules that
are recognized by the
immune system) curated
from literature and
submitted from the research
community and provides
analytical tools for epitope
data analysis and their
prediction in proteomes.
Agenda
• Introduction to basic concepts of immunological
bioinformatics.
• Overview of the Immune Epitope Database (IEDB).
• Case study #1. Prediction of peptide-MHC binding (Peters et al. A
community resource benchmarking predictions of peptide binding to MHC-I
molecules. PLoS Comput Biol. 2006 Jun 9;2(6):e65): data
compilation and
prediction methods evaluation.
• Case study #2. 3D structure based prediction of antibody
binding sites in proteins: data compilation and prediction
methods evaluation.
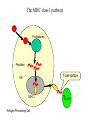
The MHC class I pathway
Antigen
Proteasome
Peptides
T-cell epitope
ER
MHC I
Antigen Presenting Cell
TCD8+
Performance measures for prediction methods
ROC curve
TP
threshold
FN
TN
sensitivity = TP / (TP + FN) =
6/7= 0.86
specificity = TN / (TN + FP) =
6/8 = 0.75
True positive rate, TP / (TP +
FN)
FP
1
0.9
0.8
0.7
0.6
AROC
0.5
0.4
0.3
0.2
0.1
0
0
0.2
0.4
0.6
0.8
False positive rate, FP / (FP + TN)
1
Prediction of MHC class I epitopes
ALAKAAAAN
ALAKAAAAV
ALAKAAAAT
GMNERPILT
GILGFVFTM
TLNAWVKVV
• Gibbs sampling
• Sequence motifs, matrices
• Sequence weighted matrices: performance of the method
(measured as AROC) depends on the number of training peptides
(”Immunological Bioinformatics” O. Lund, 2005)
0.95
• Hidden Markov Models
• Artificial Neural Networks
0.9
0.85
Aroc
ALAKAAAAM
0.8
0.75
KLNEPVLLL
AVVPFIVSV
0.7
0.65
2
10
20
100
200
Number of training peptides
Peptides
known to bind
to the HLAA*0201
molecule.
For T-cell epitopes the most selective requirement is the
ability to bind an MHC with high affinity.
500
Assembling the dataset of measured peptide affinities to MHC
class I molecules
(Peters et al. A community resource benchmarking predictions of peptide binding to
MHC-I molecules. PLoS Comput Biol. 2006 Jun 9;2(6):e65)
• Data: pairs {peptide – affinity value in terms of IC50 nM} for a given MHC
allele
• 48 different mouse, human, macaque, and chimpanzee MHC class I alleles.
• Length pf peptides 8 – 11 aa.
• If affinities for the same peptide to the same MHC molecule were recorded
in multiple assays, the geometric mean of the IC50 values was taken.
• 84% of peptides differ in at least two residues with every other peptide in
the dataset.
• 48,828 data points collected from two experimental groups.
An example of the problem of pooling experimental
data from different sources
• There is a good agreement between the
measured affinity values by two
experimental groups (Sette and Buus) for
intermediate- and low-affinity peptides, less
for high-affinity peptides.
• For peptides with high affinity of 50 nM or
better the Matthews correlation coefficient is
below 0.37.
• Important message: Pooling experimental
data from different sources requires
additional validation.
High affinity
Low affinity
(IC50 500 nM – non-binder)
Peptide binding to MHC class I affinity prediction methods comparison (the same
training and test data sets)
Correlation coefficients (ARB=0.55, SMM=0.62, ANN=0.69) are significantly different (p<0.05
using a t test).
Aroc values (ARB=0.934, SMM=0.952, ANN=0.957) are significantly different (p<0.05 using a
paired t test on Aroc values generated by bootstrap).
Peptide binding to MHC class I affinity prediction methods comparison.
Prediction performance as a function of training set size.
Peptide binding to MHC class I affinity prediction methods comparison
(external tools: different training data sets)
• Ideally the comparison should be done using ‘blind’ test set excluding
every peptide used for any method training. Otherwise the performance of
a method can be overestimated.
• That was not done in the discussed work of Peters et al.
Agenda
• Introduction to basic concepts of immunological
bioinformatics.
• Overview of the Immune Epitope Database (IEDB).
• Case study #1. Prediction of peptide-MHC binding (Peters et al. A
community resource benchmarking predictions of peptide binding to MHC-I
molecules. PLoS Comput Biol. 2006 Jun 9;2(6):e65): data
compilation and
prediction methods evaluation.
• Case study #2. 3D structure based prediction of antibody
binding sites in proteins: data compilation and prediction
methods evaluation.
HIV-1 envelope protein
gp120 core complexed
with CD4 and a
neutralizing human
antibody 17b
HIV-1 envelope
protein gp120 (core
fragment)
The entry of HIV into cells
requires the sequential
interaction of the viral exterior
envelope glycoprotein, gp120,
with the CD4 glycoprotein and
CD4 (N-terminal
a chemokine receptor on the
two domain
cell surface. These
interactions initiate a fusion of fragment)
the viral and cellular
membranes. Although gp120
can elicit virus-neutralizing
antibodies, HIV eludes the
Antibody 17b
immune system.
(Fab fragment)
17b epitope is comprised of
four discontinuous -strands.
17b epitope
PDB: 1gc1
Why is the knowledge of antibody epitopes is so important?
• Vaccine design (immunogenicity, i.e. ability of vaccine to elicit in the naïve
individual the production of pathogen neutralizing antibodies, is required):
Purified antigen (subunit) vaccines:
• Inactivated toxins “toxoids”: tetanus toxoid, diphteria toxoid
• Vaccines composed of bacterial polysaccharide antigens: flu,
pneumococcus
Synthetic antigen vaccines:
• hepatitus B (recombinant protein), herpes simplex virus
• Diagnostic design (antigenicity, i.e. ability of synthetic antigen to be recognized
by the original antibody, is required):
• Autoimmune diseases: lupus, rheumatoid arthritis
• Allergic reactions
• Basic knowledge of antigenicity.
For fusion with its target
cells, HIV-1 uses a trimeric
Env complex containing
gp120 and gp41 subunits.
There are known four
broadly reactive and
neutralizing anti-HIV mAbs
(NAbs):
b12 (epitope on gp120),
2G12 (epitope on gp120,
2F5 (epitope on gp41),
4E10 (epitope on gp41).
Other known mAbs, 44752D and 58.2 (‘V3 loop
Abs’), 17b, and X5 (‘CD4i
Abs’) have limited activity.
“HIV vaccine design and the neutralizing antibody problem” Nature Immun., 2004, 5, 233
Strategies for design immunogens that elicit broadly
neutralizing antibodies
(from “HIV vaccine design and the neutralizing antibody problem”
Nature Immunology, 2004, 5, 233)
To produce molecules that mimic the mature trimer Env on the
virion surface. These molecules can be recombinant or expressed
on the surface of particles such as pseudovirions or
proteoliposomes.
To produce Env molecules engineered to better present NAb
epitopes than do “wild-type” molecules.
To generate stable intermidiates of the entry process with the goal
of exposing conserved epitopes to which antibodies could gain
access during entry.
To produce epitope mimics of the broadly NAbs determined from
structural studies of antibody-antigen complexes.
Epitope identification
The best precision in identification of antibody epitopes is provided by X-ray
crystallography.
Other methods to predict structure and
location of antibody epitopes include:
- mass spectrometry combined with
immunoaffinity procedures;
- screening of combinatorial phagedisplay peptide libraries;
- mimitope approach: selection
ligands from a library of random
combinatorial ligands;
- alanine scan;
- etc.
Methods for antibody epitope prediction
• Sequence-based (suitable for linear epitopes only)
• Amino acid scales: hydrophobicity, secondary structure (beta-turn),
polarity, flexibility, solvent accessibility etc.
• The combination of scales and experimentation with several machine
learning algorithms showed little improvement over single scalebased methods.
• Maximum sensitivity is 59%.
• Structure-based (antibody binding site prediction for a
protein of a given 3D structure):
• CEP
• DiscoTope
• Epitope mapping using peptide libraries
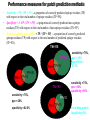
Performance measures for patch prediction methods
•
•
•
Sensitivity = TP / (TP + FN) - a proportion of correctly predicted epitope residues (TP)
with respect to the total number of epitope residues (TP+FN).
Specificity = 1- FP / (TN + FP) – a proportion of correctly predicted non-epitope
residues (TN) with respect to the total number of non-epitope residues (TN+FP).
Positive predictive value (PPV) = TP / (TP + FP) - a proportion of correctly predicted
epitope residues (TP) with respect to the total number of predicted epitope residues
(TP+FN).
TN=115
220 aa
TN=165
220 aa
FP=85
FN=5 TP=15
sensitivity = 75%,
ppv = 15%
specificity =
57.5%
FP=35
FN=5 TP=15
TN=190
sensitivity = 75%,
ppv = 30%
specificity = 82.5%
220 aa
sensitivity = 75%,
ppv = 60%
specificity = 95%
FP=10
FN=5 TP=15
For a 80aa protein
Specificity=83%
The tool purpose
Vaccine design:
Diagnostic design:
High sensitivity
High specificity and PPV
TN=200
TN=170
220 aa
TP=20 FP=30
FN=0
220 aa
TP=5
FP=0
FN=15
sensitivity = 100%
sensitivity = 25%
ppv = 40%
ppv = 100%
specificity = 85%
specificity = 100%
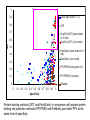
1
0.9
DiscoTope (score >-7.7)
0.8
CEP
Sensitivity
0.7
ClusPro (DOT) (best model
of 10 first)
ClusPro (DOT) (1st model)
0.6
0.5
PatchDock (best model of 10
first)
PatchDock (1st model)
0.4
0.3
0.2
PPI-PRED (best patch of 3)
0.1
PPI-PRED (1st patch)
0
ProMate
0
0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
1
Specificity
Epitope prediction methods (CEP and DiscoTope) have a tendency to be less
specific than other methods.
1
0.9
DiscoTope (score >-7.7)
0.8
CEP
0.7
ClusPro (DOT) (best model
of 10 first)
ClusPro (DOT) (1st model)
PPV
0.6
0.5
PatchDock (best model of 10
first)
PatchDock (1st model)
0.4
0.3
PPI-PRED (best patch of 3)
0.2
PPI-PRED (1st patch)
0.1
0
ProMate
0
0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
1
Specificity
Protein docking methods (DOT and PatchDock) in comparison with protein-protein
binding site prediction methods (PPI-PRED and ProMate) give better PPV at the
same level of specificity.
1
DiscoTope (score >-7.7)
0.9
CEP
0.8
ClusPro (DOT) (best model
of 10 first)
ClusPro (DOT) (1st model)
Sensitivity
0.7
0.6
0.4
PatchDock (best model of 10
first)
PatchDock (1st model)
0.3
PPI-PRED (best patch of 3)
0.2
PPI-PRED (1st patch)
0.1
ProMate
0.5
0
0
0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
1
PPV
Epitope prediction methods (CEP and DiscoTope) show worse correlation between
sensitivity and ppv than other methods (e.g. linear correlation coefficient r for CEP is 0.48,
for DiscoTope (-7.7) is 0.51, whereas r for PPI-PRED is 0.65, for CLusPro is 0,88 and
PatchDock is 0.91).