Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Genome evolution wikipedia , lookup
Minimal genome wikipedia , lookup
Gene nomenclature wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Metabolic network modelling wikipedia , lookup
Genome (book) wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Microevolution wikipedia , lookup
Gene expression programming wikipedia , lookup
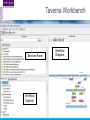
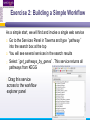
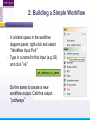
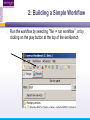
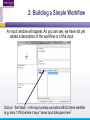
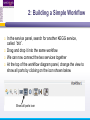
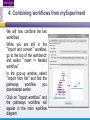
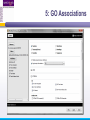
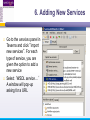
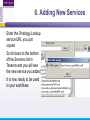
An Introduction to Designing, Executing and Sharing Workflows with Taverna and myExperiment Katy Wolstencroft University of Manchester This tutorial will give you a basic introduction to designing, and reusing workflows in Taverna and some of its main features. Workflows in this practical use small data-sets and are designed to run in a few minutes. In the real world, you would be using larger data sets and workflows would typically run for longer Exercise 1: Exploring the Workbench Taverna can be downloaded from http://www.taverna.org.uk/ Go to the page and find the latest (2.4) Follow the instructions on the website to install Taverna for your operating system (this is a simple one-click install for windows and Mac. For Linux, you may also need the GraphViz program. Follow the link on the Taverna download page if so) The following page shows a screenshot of Taverna and the different panels that make up the workbench Taverna Workbench Services Panel Workflow Explorer Workflow Diagram 1. Workflow Diagram The workflow diagram is the visual representation of the workflow, it: Shows inputs, outputs, services and data flows Allows editing of the workflow by dragging and dropping and connecting services together Enables saving of workflow diagrams for publishing and sharing 1. Workflow Explorer The Workflow Explorer shows the detailed view of your workflow. It shows default values and descriptions for service inputs and outputs and it shows where remote services are located. It also shows configuration details, such as iteration and looping Workflow validation details can also be found here. Before a workflow is run, Taverna checks to see if it is connected correctly and if its services are available. 1. Available Services Panel Lists services available by default in Taverna Local java services WSDL Web Service – secure and public RESTful Services R Processor services (for statistical analyses) Beanshell scripts Xpath scripts Spreadsheet import service The services panel also allows you to add new services or workflows from the web or from file systems – there are loads more available! 1. Taverna exercises – Enrichment Analysis 1. 2. 3. 4. Today, we will use Taverna to perform enrichment analyses on a list of genes. Many experiments result in a list of genes (e.g. microarray analysis, Chip-Seq, SNP identification etc). In this case, the genes are those associated with ChipSeq peaks We will enrich our dataset by discovering: Which pathways our genes are involved in The functions of the genes The cellular locations of the gene products Literature evidence for the phenotype/trait of interest Exercise 2: Building a Simple Workflow As a simple start, we will find and invoke a single web service Go to the Services Panel in Taverna and type ‘pathway’ into the search box at the top You will see several services in the search results Select ‘get_pathways_by_genes’. This service returns all pathways from KEGG Drag this service across to the workflow explorer panel 2: Building a Simple Workflow In a blank space in the workflow diagram panel, right-click and select “Workflow Input Port” Type in a name for this input (e.g. ID) and click “ok” Do the same to create a new workflow output. Call this output “pathways” 2: Building a Simple Workflow You now have 3 boxes in the diagram and we need to connect them up Click on the input box and drag towards “get_pathways_by_genes” and let go. An arrow will connect the two boxes 2: Building a Simple Workflow Click on the output box, drag towards “get_pathways_bygenes”, and let go. A pop-up will ask you to select from ‘attachmentList’ and ‘return’. Select ‘return’ An arrow will connect the two boxes You have now built your first workflow! It should look something like this 2: Building a Simple Workflow Run the workflow by selecting “file -> run workflow”, or by clicking on the play button at the top of the workbench 2: Building a Simple Workflow An input window will appear. As you can see, we have not yet added a description of the workflow or of the input Click on ‘Set Value’ in the input window and add a KEGG Gene identifier (e.g. mmu:13163) where it says “some input data goes here” 2: Building a Simple Workflow Click “run workflow”. You will automatically be switched to the ‘Results’ window. Taverna will run the KEGG Web Service in Japan to return pathways that gene is involved in. In the bottom left of the results window, click on the results. You will see some pathway identifiers. These are good for computers, but not for humans. We need pathway descriptions to properly examine the results Switch back to the ‘Design’ window using the tab at the top of the workbench 2: Building a Simple Workflow In the service panel, search for another KEGG service, called ‘btit’. Drag and drop it into the same workflow We can now connect the two services together At the top of the workflow diagram panel, change the view to show all ports by clicking on the icon shown below Show all ports icon 2: Building a Simple Workflow Connect the get_pathways_by_gene ‘return’ output to the input (string) of btit Create a new output called ‘pathway_description’ and connect it the btit ‘return output by dragging an arrow between them Re-run the workflow and look at the pathway descriptions The workflow will iterate over each pathway ID to find each description 2: Building a Simple Workflow A list of pathways and their descriptions is useful, but it would be easier to visualise diagrams of the whole pathways Additionally, we need to find ALL the pathways for ALL the genes in our lists in order to indentify which pathways are over-represented in our data set For both these tasks we will find and use workflows from myExperiment Exercise 3: Re-using workflows from myExperiment Go to http://www.myexperiment.org and click on ‘find workflows’ You will see a list of the most viewed and downloaded workflow – see what the most popular workflow does by reading the description Change the rank to ‘Latest’ and see what has been uploaded in the last few weeks 3: Re-using workflows from myExperiment Find the workflow called “geneID to KEGG Pathways” and look at the workflow entry page (note: if your search returns too many results, you can refine it by adding “Wolstencroft” Download the workflow by clicking on the link: “Download Workflow File/Package (T2FLOW)” Open the workflow in Taverna by going to ‘File ->Open Workflow’ Run the workflow using the example values supplied by the workflow creator (Hint: when you run the workflow the examples values will be added by default in the input window) Look at the workflow output – now you will see pathway diagrams Exercise 4: Combining workflows from myExperiment To analyse all the genes from our study, we need to extract the gene list from previous analysis results To make it easier to work through the example, we have provided a Chip-Seq gene list on myExperiment: http://www.myexperiment.org/files/661.html Save this file to your local machine Open the file in Excel Save the file with a .csv extension As you can see, the list of genes is in column D Taverna can process and extract this column automatically 4: Combining workflows from myExperiment In myExperiment, find and download the workflow called “Import and convert gene list” This workflow will extract the list of genes in column D using Taverna’s built-in spreadsheet import tool (which can be found in the services panel, for future reference) The next step in the workflow converts the RefSeq IDs into unigene IDs (required for the pathways workflow – converting between different types of identifiers is a common problem in bioinformatics!) Run the workflow. This time, in the input window, select “set file location” and set the location to the saved .csv gene list. Look at the workflow results 4: Combining workflows from myExperiment We will now combine the two workflows While you are still in the “import and convert” workflow, go to the top of the workbench and select “ insert -> Nested workflow” In the pop-up window, select “import from file” and find the pathways workflow you downloaded earlier. Click on “import workflow” and the pathways workflow will appear in the main workflow diagram. 4: Combining workflows from myExperiment Connect the workflows up by linking the output of the ‘Merge_Gene_List’ with the nested workflow input 4: Combining workflows from myExperiment Create new output ports for the Nested workflow and connect the Nested workflow outputs to the new outputs NOTE: you don’t need to connect them all, just pathway descriptions, pathway images and gene descriptions Save the workflow Run the workflow 4: Combining workflows from myExperiment The workflow may take a few minutes to run. Spend the time looking at myExperiment to find other pathway-related workflows What other pathway workflows are there? Do they all use KEGG? What other resources could you use instead? Exercise 5: GO Associations There are many different tools we could use to find Gene Ontology associations for your gene list For example, we could simply modify the BioMart/Ensembl service in the ‘Import and convert gene list’ workflow we have already used Reload the ‘Import and Convert gene list’ workflow Right-click on the ‘mmusculus_gene_ensembl’ service and select ‘Copy’ Paste an extra copy of this service into the same workflow diagram 5: GO Associations This is a BioMart service. It allows you to retrieve omics data from ENSEMBL and other genomics resources. If you are familiar with BioMart, you will see the interface in Taverna is very similar to the web interface We will modify the BioMart query to find all GO associations for each gene associated with a Chip-Seq peak Right-click on the new copy of the service and select ‘Configure BioMart Query’ 5: GO Associations The inputs (or filters) already accept RefSeq Ids from our input file, but we need to modify the outputs (or attributes) Select ‘Attributes’ and expand the ‘External’ section. Unselect ‘UniGeneID’ and select ‘RefSeq mRNA’ Additionally, select ‘Go Term Accession’, ‘GO Name’ and ‘Go Domain’ At the top of the page, change the output format from multiple to single (TSV format) (See screenshot on the next slide for an example) 5: GO Associations 5: GO Associations Click ‘apply’ to save your changes, and ‘close’, to go back to Taverna At the top of the workflow diagram, change the workflow view to show all ports by clicking on the table icon 5: GO Associations Connect your new service to the workflow by linking the ‘D’ output port of the spreadsheet service to the input of your new service Make a new output port called ‘GO_Report’ and connect it to your new service 5: GO Associations Save the workflow by going to ‘File -> Save Workflow’ Run the workflow Download and view the GO report Exercise 6: Adding New Services to Taverna In Taverna, new tools can be ‘added’ very easily because we are often actually calling external tools Go to http://www.biocatalogue.org and look around. Biocatalogue is a registry of available Web Services for the Life Sciences. You can use any of these tools in Taverna Search for the ‘ontology lookup service’ Look at the entry for that service find and copy the WSDL location URL HINT: it will be a URL ending in .wsdl (http://....wsdl) 6. Adding New Services Go to the services panel in Taverna and click “import new services”. For each type of service, you are given the option to add a new service Select ‘WSDL service…’ A window will pop-up asking for a URL 6. Adding New Services Enter the Ontology Lookup service URL you just copied Scroll down to the bottom of the Services list in Taverna and you will see the new service you added It is now ready to be used in your workflows 6: Adding New Services to Taverna Now we have Gene Ontology descriptions for our genes, we might want to find out what other ontology descriptions we can find From the service set you have just imported, add the service ‘getontologyname’ to a new workflow This service does not require any inputs, so just create an output port called ‘ontologyNames’ and connect it to the service Run the workflow You will see a list of all ontologies you can search using these services Sometimes, documentation about services is embedded in the service set like this Exercise 7: Text Mining So far we have looked at enriching the genomic information, but we could also use workflows for running data analyses (e.g. aligning mouse genes with human homologues) or performing literature searches Think about the ways you could extend this analysis with literature searches (e.g. Correlations between pathways, genes, GO terms, phenotypes etc) Search myExperiment for workflows involving text mining, using the search terms “text mining” and “Pubmed” 7: Text Mining Find and open the workflow “Phenotype to pubmed” One of the services is no longer available in the nested workflow (the faded-out service). Taverna checks the availability of each service when you load the workflow and when you run it In this case, the workflow will still run without the final nested workflow (clean text) Delete the ‘clean text’ nested workflow (by selecting it and right-clicking), and reconnect the workflow output Run the workflow with the search term ‘erythropoiesis’ (or a phenotype term to describe the disease you are studying) 8: Sharing Workflows If you want to save and share any workflows on myExperiment, you can create an account and upload them If you wish to share them with each other, we can set up a workshop group with restricted membership 8: Outcomes These exercises have given you a brief introduction to Taverna, but we have just scratched the surface. The examples are taken from a real investigation, but the data has been reduced to a level that will run in a few minutes. If you would like to know more about using particular types of services, for example REST, or R, or the External tools plugin, we have other tutorial material. We also have material to explain the advanced engine features, such as iteration, looping, parallel invocation and retries.