Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Protein moonlighting wikipedia , lookup
Human genome wikipedia , lookup
Genetic code wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Point mutation wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Metagenomics wikipedia , lookup
Helitron (biology) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Pattern Discovery
in Biological Sequences:
A Review
ChengXiang Zhai
Language Technologies Institiute
School of Computer Science
Carnegie Mellon University
Presentation at the Biological Language Modeling Seminar, June17, 2002
Outline
Algorithm
Application
Computer
Science
Pattern
Discovery
Biology
Motivation
Formalization
Basic Concepts (“Common Language”)
Basic Concepts
• Alphabet & Language
– Alphabet = set of symbols, e.g., ={A, T, G, C} is
the nucleotide alphabet
– String/Sequence (over an alphabet) = finite seq. of
symbols, e.g., w=AGCTGC ( How many different
nucleotide strings of length 3 are there?)
– Language (over an alphabet) = set of strings, e.g.,
L={AAA, AAT, ATA, AGC, …, AGG} all nucleotide triplets starting
with A.
Example:“Essential AA Language”
The language (set) of “essential” amino acids
on the alphabet {A, U, C, G}
L={CAC, CAU, …, UAC, UAU}
The Genetic Code
Questions to Ask about
a Language (L)
• Syntax & Semantics
– How do we describe L and interpret L?
• Recognition
– Is sequence s in L or not?
• Learning
– Given example sequences in L and not in L, how do
we learn L? What if given sequences that either
match or do not match a sub-sequence in L ?
Syntax & Semantics of Language
• Syntax: description of the form of sequences
– “Surface” description: enumeration
– “Deep” description: a concise decision rule or a
characterizing pattern, e.g.,
• L contains all the triplets ending with “A”, or
• L contains all sequences that match “AGGGGGA”
• Semantics: meaning of sequences
– Functional description of a amino acid sequence
– Gene regulation of a nucleotide sequence
Recognizing Sequences in L
• Recognizer (for L): given a sequence s, it tells
us if s is in L or not. An operational way of
describing L!
L (G-receptors)
Algorithm
(G-rec. Recgonizer)
*
( all protein sequences)
Is the sequence “SNASCTTNAP…TGAK” a G-receptor?
0 (no)
1 (yes)
More than “recognizing”...
• Can the recognizer explain why a sequence is
a “G-receptor”? Is the explanation
biologically meaningful?
• The “explanatory power” reflects the
recognizer’s understanding of the language.
• Two possible explanations/decision rules:
– It is longer than 300 AA’s
– The four AA’s “A, P, K, B” co-occur within a
window of 50 AA’s
Learning a Language
(from Examples)
Positive examples
L
*
Negative examples
Learn a recognizer (Classification)
- Given a new sequence, decide if it is in L
Learn meaningful features (Feature Extraction/Selection)
- Characterize, in a meaningful way, how L is different
from the rest of *
More Basic Concepts
• Pattern/Motif sequence template, e.g., A..GT
• Different views of a pattern
– A pattern defines a language: L={ seq’s that match
the pattern} =>
• Language learning ~ pattern learning?
• Given a language, can we summarize it with a pattern?
– A pattern is a feature: The feature is “on” for a
sequence that matches the pattern =>
• Feature extraction ~ pattern extraction?
– A pattern is a sequence of a pattern language
The Need of Probabilities
• We have many uncertainties due to
– incomplete data and knowledge
– noise in data (incorrectly labeled, measurement
errors, etc)
• So, we relax our criteria
– L could potentially contain all the sequences, but
with different probabilities (“statistical LM”)
– How likely is a sequence s in L?
– How do we learn such an L? (LM estimation)
Biological Motivation for
Pattern Discovery
• Motifs or “preserved sequence patterns” are
believed to exist
• Motifs determine a sequence’s biological
function or structure
• Many successful stories (Brejova et al. 2000)
– Tuberculosis: detecting “secretary proteins” 90%
confirmed
– Coiled coils in histidine kinases: detecting coiled
coil
Amino Acid Patterns:
Patterns in Protein
• Possible biological meanings: They may
– determine a protein’s 3-D structure
– determine a protein’s function
– indicate a protein’s evolutionary history or family
• “Suspected” properties: They may be
– long and with “gaps”
– flexible to permit substitutions
– weak in its primary sequence form
– strong in its structural form
Nucleotide Patterns I:
non-coding regions
• Possible biological meanings: They may
– determine the global function of a genome, e.g.,
where all the promotors are
– regulate specific gene expression
– play other roles in the complex gene reg. network
• “Suspected” properties: They may be
– in the non-coding regions
– relatively more continuous and short
– working together with many other factors
Nucleotide Patterns II:
Patterns in RNA
• Possible biological meanings: They may
– determine RNA’s 3-D structure, thus indirectly
transcription behavior
• “Suspected” properties: They may
– be long and with “gaps”(?)
– contain many “coordinating/interacting” elements
– weak in its primary sequence form
– strong in its structural form
Nucleotide Patterns III:
Tandem Repeats
• Possible biological meanings: They may
– be a result of mutations from the same original
segment
– play a role in gene regulation
– be related to several diseases
• “Suspected” properties: They may
– be contiguous
– approximate copies of the same “root form”
– be hard to detect
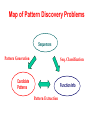
Pattern Discovery Problem Formulation
• The ultimate goal is to find Meaningful
Patterns
• Broadly three types of sub-problems
– Pattern Generation/Enumeration
– Sequence Classification/Retrieval/Mining
– Pattern Extraction
Map of Pattern Discovery Problems
Sequences
Pattern Generation
Seq. Classification
Candidate
Patterns
Function Info
Pattern Extraction
Pattern Generation/Enumeration
• Given a (usually big) collection of sequences
• Generate/enumerate all the “significant”
patterns that satisfy certain constraints
• Issues:
– Design a pattern language (e.g., max. length=?)
– Design significance criteria (e.g., freq >= 3)
– Design a search/enumeration strategy
– Algorithm has to be efficient
Sequence Classification
• Finding structures on a group of sequences
– Categorization: group sequences into known
families
– Clustering: explore any natural grouping tendency
– Retrieval: find sequences that satisfy certain need
• Goal: maximize classification accuracy
• Issues:
– Dealing with noise + Using good features/patterns
– Breaking the limit of “linear similarity”
Sequence Categorization
• 2 or more meaningful classes
• Examples available for each class
• Goal is to predict the class label of a new
instance as accurately as possible
• E.g., protein categorization, G-receptor
recognition
Learn the boundaries
Examples
C3
C2
C1
Sequence Clustering
• Given sequences, but no pre-defined classes
• Design similarity criteria to group sequences that
are similar
• Goal is to reveal any interesting structure
• E.g., gene clustering based on expression
information
Learn the boundaries
Sequence Retrieval
• Given some desired property of sequences
• Find all sequences with the desired property
• E.g., find all Enzyme sequences that are similar
to my G-receptor sequence
Query
Find these sequences
Pattern Extraction
• Suppose you are given a lot of text in a foreign
language unknown to you
• Can you identify proper names in the text?
• Issues:
– Need to know the possible form of a meaningful
pattern (Will a name have more than three words?)
– Need to identify useful “clues” (e.g., Capitalized)
– The extraction criteria must be based on some
information about the functions or structures of a
sequence
Entering the Algorithm Zone ...
• The most influential ones seem to be:
– Pattern Generation Algorithms
• TEIRESIAS & SPLASH
– Pattern Classification Algorithms
• I believe that most standard classification algorithms have
been tried
• HMMs are very popular
– Pattern Extraction Algorithms: ???
TEIRESIAS & SPLASH
• Find deterministic patterns (exact algorithm)
• Pattern Language:
– Allowing gaps, e.g. A..H…C
– Constraints on density of the wild-card “.”
– Less powerful than the regular language/expression
• Significance criteria
– Longer = more significant
– Higher frequency = more significant
– Statistical test: How likely is it a random effect?
Basic Idea in TEIRESIAS & SPLASH
• Generate & Test
• Pruning strategy: If a (short) pattern occurs
fewer than 5 times, so do ALL longer patterns
containing it!
• A “Bottom-up” Inductive Procedure
– Start with high frequency short patterns
– At any step, try to extend the short patterns slightly
Possible Applications of
TEIRESIAS & SPLASH
• Defining a feature space (“biological words”)
• Suggesting structures for probabilistic models
(e.g. HMM structure)
• A general tool for any sequence mining task
(e.g., mining the web “click-log” data?)
Map of Pattern Discovery Problems
Pattern Meaningful Structure?
Sequences
Structure Analysis
(Alignment)
Structure Info
Function Analysis
(Classification)
Function Info
Pattern Extraction/Interpretation
Probabilistic Pattern Finding
• Probabilistic vs. Deterministic Patterns
– Functional comparison
• A deterministic pattern either matches or does not match a
sequence
• A probabilistic pattern potentially matches every sequence,
but with different probabilities
• Deterministic patterns are special cases of prob. Patterns
– Structural comparison
• Deterministic patterns are easier to interpret
Hidden Markov Models (HMMs)
• Probabilistic Models for Sequence Data
• The “System” is in one of K states at any
moment
• At the next moment, the system
– moves to another state probabilistically
– outputs a symbol probabilistically
• To “generate” a sequence of n symbols, the
system makes n “moves” (transitions)
Examples
1.0
Unigram LM
P(w1…wn) = p(w1|s)…p(wn|s)
= p(w1)…p(wn)
s
p(w|s)
Position Weight
Matrix(PWM)
Start
1.0
1.0
s1
p(w|s1)
s2
1.0
...
p(w|s2)
sk
1.0
End
p(w|sk)
P(w1…wn) = p(w1|s)…p(wn|s)
Deterministic Pattern
AUG……[UAG|UGA|UAA]
1.0
Start
A
U
G
1
2
3
p(“A”)=1 p(“U”)=1 p(“G”)=1
p(“A”)+ p(“U”)+ p(“G”) + p(“C”)=1
U
A
A
G
A
A
G
End
Three Tasks/Uses
• Prediction (Forward/Backward algorithms)
– Given a HMM, how likely would the HMM generate a
particular sequence?
– Useful for, e.g., recognizing unknown proteins
• Decoding (Viterbi algorithm)
– Given a HMM, what is the most likely transition path for
a sequence (discover “hidden structure” or alignment)
• Training (Baum-Welch algorithm)
– HMM unknown, how to estimate parameters?
– Supervised (known state transitions) vs. unsupervised
Applications of HMM
• MANY!!!
• Protein family characterization (Profile HMM)
– A generative model for proteins in one family
– Useful for classifying/recognizing unknown proteins
– Discovering “weak” structure
• Gene finding
– A generative model for DNA sequences
– Identify coding-regions and non-coding regions
An Example Profile HMM
• Three types of states:
delete
– Match
– Insert
– Delete
insert
• One delete and one
match per position in
model
• One insert per
transition in model
• Start and end “dummy”
states
alignment
Match (at position 2)
Example borrowed from Cline, 1999
Profile HMMs: Basic Idea
• Goal: Use HMM to represent the common
pattern of proteins in the same family/domain
• First proposed in (Krogh et al. 1994)
• Trained on multiple sequence alignments
– #match-states = #consensus columns
– Supervised learning
• Trained on a set of raw sequences
– #match-states = avg-length
– Unsupervised learning
Uses of Profile HMMs
• Identify new proteins of a known family
– Match a profile HMM with a database of sequences
– Score a sequence by likelihood ratio (w.r.t. a
background model), apply a threshold
• Identify families/domains of a given sequence
– Match a sequence with a database of profile HMMs,
– Return top N domains
• Multiple alignments
• Identify similar sequences: Iterative search
Profile HMMs: Major Issues
• Architecture: Explain sub-families, more
constrained (motif HMMs)
• Local vs. global alignment
• Avoid over-fitting: Mixture Dirichlet prior, Use
labeled data
• Avoid local-maxima: Annealing, labeled data
• Sequence weighting: Address sample bias
• Computational efficiency
Profile HMMs: More Details
• Dirichlet Mixture Prior
– Generate an AA from a Dirichlet distribution Dir(p|) in
two-stages
– Given observed AA counts, we can estimate the prior
parameters ’s
– Assume a mixture of k Dirichlet distributions Dir(p|)
– For each column of multiple alignment
– Assume that the counts (of different AA’s) are a
sample of the mixture model
Protein Structure Prediction
with HMMs
• SAM-T98:
– Best method that made use of no direct structural
information at CASP 3 (Current Assessment of
Structure Prediction)
– Create a model of your target sequence
– Search a database of proteins using that model
– Whichever sequence scores highest, predict that
structure
How do we build a model using
only one sequence?
Application Example:
Pfam (HMMER)
•
Pfam is a large collection of protein multiple sequence alignments and profile
hidden Markov models. Pfam is available on the World Wide Web in the
UK,…, Sweden, …, France, …, US. The latest version (6.6) of Pfam contains
3071 families, which match 69% of proteins in SWISS-PROT 39 and TrEMBL
14. Structural data, where available, have been utilised to ensure that Pfam
families correspond with structural domains, and to improve domain-based
annotation. Predictions of non-domain regions are now also included. In
addition to secondary structure, Pfam multiple sequence alignments now
contain active site residue mark-up. New search tools, including taxonomy
search and domain query, greatly add to the functionality and usability of the
Pfam resource.
HMM Gene Finders
• Goal: Use HMM to find the exact boundary of
genes
• Usually Generalized HMMs
– “With Class” (GeneMark & GeneMark.hmm?)
– State = Neural Network (Genie)
• Architecture: 2 modules interleaved
– Boundary module: start codon, stop codon, binding
sites, transcription factors, etc.
– Region module: exons, introns, etc.
– A lot of domain knowledge encoded
HMMs: Pros & Cons
Advantages:
Disadvantages:
– Statistics
– State independence
– Modularity
– Over-fitting
– Transparency
– Local Maximums
– Prior Knowledge
– Speed
More Applications & Discussions
• Ultimately how useful are these algorithms
for biology discovery?
– Integrated with biological experiment design
(reinforcement learning?)
– Biological verification of patterns/classification
• Evaluation of these algorithms is generally
hard and expensive?
Some Fundamental Questions
• How powerful should the pattern language
be? Is “regular expression” sufficient?
• How do we formulate biologically meaningful
or biologically motivated
classification/extraction criteria?
• How do we evaluate a pattern without
expensive biological experiments?