Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
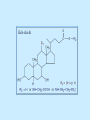
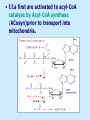
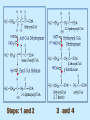
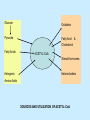
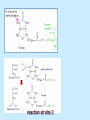
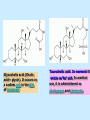
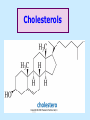
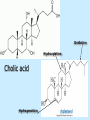
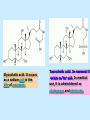
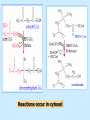
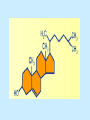
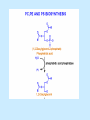
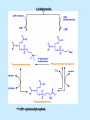
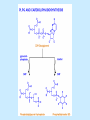
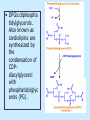
BIOCHEMISTRY SFA 2073 Lipid Metabolism NIK NORMA NIK MAHMOOD-Ph.D FACULTY OF SCIENCE AND TECHNOLOGY ISLAMIC SCIENCE UNIVERSITY OF MALAYSIA Digestion & Absorption • Lipids that gets into the digestive system are dietary lipids, normally free fatty acids, cholesterols and triglycerides (TAG) and many minor components. • Digestion starts in the duodenum portion of small intestine: - is mix with bile that contains HCO3ˉ ions and bile salts to solubilize fat. This process is called emulsification. The big lipids droplets are broken down into smaller droplets. (bile is made in liver and stored in gall bladder between meals. When there is food, bile is delivered to the intestine from gall bladder via bile duct) - TAG is acted by lipase secreted by pancreas liberating monoglyceride and two fatty acids. Monoglyceride, cholesterol and f.fas and bile salts form amphipathic micelles. These micelles keep the insoluble lipid components in soluble aggregates from which small amounts are released and absorbed by epithelial cells via diffusion. - Free fatty acids and monoglycerides then recombine into triacylglycerols at the smooth ER and together with cholesterols moves on to Golgi to be converted to chylymicrons. It enters interstitial fluid, then taken up by the lacteals in the intestinal wall and delivered to liver via hepatic portal vein for processing. • exposure to a large aggregate of triglyceride, the hydrophobic portions of bile acids intercalate into the lipid, with the hydrophilic domains remaining at the surface. Such coating with bile acids aids in breakdown of large aggregates or droplets into smaller and smaller droplets. • Pancreatic lipases hydrolyse triglyceride into monoglyceride and free fatty acids. The activity of this enzyme is clipping the fatty acids at positions 1 and 3 of the triglyceride, leaving two free fatty acids and a 2-monoglyceride. **Lipase is a water-soluble enzyme • Lipids, and products of their digestion are transported through aqueous compartments within the cell as well as in the blood and tissue spaces in the forms LIPOPROTEINS Why in the form of LIPOPROTEINS? • Large portion of the lipids’ structures comprise of C-C &C-H rendering lipids hydrophobic in nature i.e lipids are insoluble in aqueous environment thus creates problem to its transport within bodymedium which is aqueous in nature. Dietary triacylglycerols (Tag) & cholesterol and in-vivo Tag and cholesterol (synthesized in liver), must be converted to the soluble form to overcome the problem. This is achieved by forming LIPOPROTEINS Lipolysis • Is the breakdown of fat (Tag) stored in fat cells into free fatty acids + glycerol + mono & diglycerides which is catalysed by enzyme lipase • Induced by hormone epinephrine , norepinephrine, glucagon and adreno corticotropic hormone • the lipolytic products are then released into the blood • The free fatty acids bind to serum albumin and transport to tissues that require energy. The energy is generated by catabolic β-oxidation pathway (a 4 steps pathway/cycle) • How the hormones induce lipolysis ? The hormones trigger 7TM receptors, which activate adenylate cyclase. This results in increased production of cAMP, which activates protein kinase A, which subsequently activate lipases found in adipose tissue. • β–oxidation of free fatty acid Fatty acid degradation and synthesis are relatively simple processes and essentially the reverse of each other. • f.f.a first are activated to acyl-CoA catalyse by Acyl-CoA synthase (ACosyn)prior to transport into mitochondria. • acylCoA not permeable to inner mitochondria membrane, hence carried across by carnitine carrier system into mitochondrion matrix. It is by conjugation to carnitine. AcylCoA + Carnitine → Acyl-carnitine + Co-ASH • carnitine carrier system consisted of 2 enzyme-units CAT I & CAT II Carnitine Acyl Transferase Carnitine (a quaternary ammonium compound) is hydrophilic amino acid derivative, produced endogenously in the kidneys and liver from lysine and methionine of diet’s meat and dairy products. Carnitine binds acyl residues conjugated with coenzyme A. Carnitine + Acyl-CoA Inner membrane + CoASH matrix CAT II Acyl-carnitine Acyl-CoA Intermembrane space ACosyn cytosol CAT I + CoASH Outer membrane Fatty acid CAT: carnitine Acyl transferase Simplified mitochondrial layout ACosyn: acyl-CoA synthetase • Followed by (4 steps ) • - oxidation by FAD • - hydration • - oxidation by NAD+ • - thiolysis • The cycle then repeats on the larger fragment while acetyl-CoA fragment channeled to Krebs Cycle Step 1 • oxidation by FAD/Acyl-CoA DH : The activated fatty acid is oxidized to introduce a double bond. Step 2 • Hydration/Enoyl-CoA Hydratase: to introduce an oxygen via formation of alcohol Step 3 • oxidation by NAD+/Hydroxy-CoADH: the alcohol is oxidized to a ketone. Step 4 • Thiolysis- Thiolase/CoA-SH : cleaving of the acylCoA into two fragments, acetyl CoA and an acylCoA of fatty acid chain two carbons shorter. Steps: 1 and 2 3 and 4 • β-oxidation of unsaturated fatty acids poses a problem. Unsaturated f.a are the cis type. This prevents the formation of the required bond orientation, trans-δ2 bond, in the enoyl intermediate. These situations are handled by an additional of two enzymes. Anabolism Of Fatty Acid in Human • Process occurs in cytoplasm of liver (major) and adipose tissue cells. • Fatty acids are formed by the following 3 rxn-stages: i- acetyl-CoA Carboxylase rxn ii- fatty acid synthase rxn iii- desaturase rxn • This process is the de novo synthesis of F.A • The initiator substrate acetyl-CoA is the product of β-oxidation catabolic pathway Cytoplasm Mitochondria Acetyl CoA Acetyl CoA synthesis β-oxidation citrate citrate Fatty Acid oxaloacetate Fatty Acid NADH oxaloacetate Citrate synthase NADH malate malate Malate DH ATP-citrate lyase NAD+ NADP+ NAD+ Malic enzyme NADP+ NADPH pyruvate NADPH pyruvate transporter Malate-oxaloacetate shuttle: Transfer OF Acetyl CoA from Mitochondria to cytoplasm Glucose Oxidation Pyruvate Fatty Acid & Cholesterol Fatty Acids ACETYL-CoA Steroid hormones Ketogenic Ketone bodies Amino Acids SOURCES AND UTILIZATION OF ACETYL-CoA Acetyl-CoA Carboxylase rxn • Initiator to fatty acid synthesis is acetyl-CoA • Acetyl-CoA carboxylase catalyses carboxylation of acetyl-CoA to malonyl-CoA via 2-steps reaction. • The enzyme is biotin bound. In mammals acetyl-CoA carboxylase is a large enzyme controlled by conversion inactive ══> active (inactive: protomers (4 subunits; one biotin); active: 1 unit) conversion promoted by citrate, but inhibited by fatty acyl CoA. Also by hormonal controlled: in liver by glucagon – PO4rylation to inactive form; in adipose tissue by adrenalin (epinephrin) – PO4rylation to inactive form Additional note • Acetyl-CoA originated from pyruvate in mitochondria and transported to cytosol as citrate by condensing with oxaloacetate • In cytosol citrate is broken down to yield acetyl-CoA and oxaloacetate by ATP-citrate lyase. • Acetyl-CoA undergoes carboxylation by Acetyl-CoA carboxylase to malonyl-CoA Figure at right is expansion of reaction in figure on left reaction at site 2 • ATP-dependent carboxylation of the biotin, carried out at one active site (1) • transfer of the carboxyl group to acetyl-CoA at a second active site (2). • Reaction is spontaneous, HCO3- + ATP + acetyl-CoA → ADP + Pi + malonyl-CoA Fatty acid synthase rxn • The reaction is a multi-steps . • The enzyme(in mammal) is a very large polypeptide of many domains that includes an acyl carrier protein domain. • Has a number of prosthetic grps. • Individual domain catalyses a single step . • the precursor of fatty acid synthesis is malonylCoA • the initial action is binding of acetyl-CoA (2C) and malonyl-CoA (3C) to specific domain of FAS leading to formation of acyl-ACP intermediates 5C (steps 1&2) • Followed by formation of βketoacyl-ACP (4C) with evolution of CO2 (step 3) • β-ketoacyl is reduced to an alcohol, by electron transfer from NADPH (step 4). • Dehydration yields a trans double bond (step 5). • Reduction at the double bond by NADPH yields a saturated Acyl-ACP chain- 4C (step 6). This is 1 cycle. • Acyl-ACP and malonyl ACP then repeat step 3 and reaction proceeds to step 7. • Acyl chain lengthens by 2C / cycle. • Elongation process stops when acyl 16C is formed. Hydrolysis of the ester bond takes place with liberation of palmitate. FAS **The active enzyme is a dimer of identical subunits. All of the reactions of fatty acid synthesis are carried out by the multiple enzymatic activities of fatty acid synthase FAS REGULATION of f.f a synthesis • The major site of fatty acid synthesis regulation is at reaction catalysed by acetyl-CoA carboxylase (ACC). ACC requires a biotin co-factor Activity of ACC is associated with conformational change of the enzyme, and conc. of citrate and palmitoyl-CoA. When [citrate] is high, monomeric form associates to the multimeric form. Active conformation is the multimeric form. When [palmitoyl] is high, multimeric form dissociates into monomeric,it becomes inactive. citrate n monomeric –PO4 inactive (multimeric)n + Pi active palmitoyl Catabolism Of Triglycerides (TG) • Initial rxn is in small intestine where TG is mixed with bile salt. • Bile salt are steroids with detergent properties. 2 most abundant componants are cholate and deoxycholate, and they are normally conjugated with either glycine or taurine Glycocholic acid (Cholic acid+ glycin). It occurs as a sodium salt in the bile of mammals Taurocholic acid. In mammal it exists as Na+ salt. In medical use, it is administered as cholagogue and choleretic. • Starts by break-up of the glyceride (TG) into fatty acids and monoacylglycerol by pancreatic lipases. This step takes place because TG cannot be transported across the plasma membrane of the intestinal wall cells (enterocytes) due to its size. • The 2 products are transported into the cell. Once in the cell, recombination occurs and triacylglyerols TG1 are reformed. • TG1 is combined with dietary cholesterol, newly synthesized phospholipids and protein into compound chylomicrons (a large, low-density lipoproteins). • Lipoprotein lipase (synthesize by a number of sources) acts on TG1 portion in the chylomicron liberating F.F.A and glycerol. F.F.A is metabolised by either: - converted to new TG - catabolic pathway(β-oxidation) - used in membrane synthesis • The glycerol is transported to and absorbed by the liver or kidney where it is converted to glycerol-3phosphate by the enzyme glycerol kinase,GK. Glycerol 3-phosphate (especially from hepatic) converted into dihydroxyacetonephosphate (DHAP) then glyceraldehyde-3phosephate(G3P) to join glycolysis and gluconeogenesis pathway. IN INTESTINE TG1 in chylomicron Lipoprotein lipase F.F.A Glycerol Glycerol kinase TG Membrane synthesis Catabolism β-oxidation Glycerol-3-PO4 TG phospholipids Glucose Anabolism Of Triglyceride • Precursor is L-glycero-3phosphate • Proceed by condensation with acyl-CoA to form lysophosphatidic acid (l.p.a), catalyse by enzyme E1 PEP 1 L-glycerol-3-PO4 2 Glycerol *1 is glycerol-3-PO4 DH ; 2 is glycerol kinase *E1 is glycerol-3-PO4 acyltranferase Further reactions on l.p.a till formation of TG Cholesterols CHOLESTEROL • Is a soft, fat-like, waxy substance found in the bloodstream and membrane of cells (especially of the liver, spinal cord), and myelin sheaths and some hormones. • require by cells as a precursor to bile acids. • it is transported in the circulatory system within lipoproteins. • The most abundant of the steroids **Steroids are complex derivatives of triterpenes They are characterized by a carbon skeleton consisting of four fused rings. • normal adult utilized ~1 gram of cholesterol daily. Approximately 70% of the amount produces by the liver. The other 30% comes from dietary intake • Cholesterol is the precursor for all steroids. It is a common component of animal cell membranes and functions to help stabilize the membrane. Thus it is a crucial molecule *high levels of it in the blood may contribute to atherosclerosis. Catabolism Of Cholestrols • Is not the usual mode i.e brokendown to smaller molecules • Instead it is converted to the more soluble derivatives to facilitate its degradation and excretion. • Most important mechanism is the formation of bile acids, in liver. • Bile Acids (BA) are mixtures of compounds and possess digestive function as agent for emulsification and absorption of dietary fats. BA are important component of bile. Cholic acid and deoxycholate are 2 of the components of BA. Metabolism of Cholesterol cholesterol 7-α-hydroxycholestrl Many2 steps glycocholate Cholic acid (-ve charge) glycine (-ve charge) Oxidation Hydroxylation Cholic acid Hydrogenation Usually the BA are converted to a more soluble form by conjugation with glycine or taurine glycine /NH2CH2CO2H taurine (an a.a)/NH2CH2CH2SO3H e.g Conjugation: cholic acid + Glycine → glycocholate cholic acid + taurine → taurocholic acid Glycocholic acid. It occurs as a sodium salt in the bile of mammals Taurocholic acid. In mammal it exists as Na+ salt. In medical use, it is administered as cholagogue and choleretic. • Regulation of cholesterols level in blood -Absorbed dietary cholesterol increased linearly with the increase of dietary cholesterol intake. - The higher the fractional and absolute absorption of dietary cholesterol the lower the rates of biliary secretion, fecal elimination, and cholesterol synthesis (regulate cholesterol elimination and synthesis). - high serum levels of total, LDL, and HDL cholesterol were associated with high cholesterol absorption Anabolism Of Cholesterol • Condensation of precursors, acetylCoA and acetoacetyl-CoA catalyse by Hydroxymethyl glutaryl CoA synthase (HMG-CoA synthase). • Reactions proceed to formation of Mevalonate, catalysed by HMG-CoA Reductase . This rxn is rate-limiting Reactions occur in cytosol • To this structure other rings are added to form the final product • Enzyme HMG-CoA Reductase is highly regulated and the target of pharmaceutical intervention. Regulation of Cholesterol Synthesis: • is not direct on cholesterol but through B.A • B.A is removed from pool by dietary fibers • Depletion of B.A induces synthesis of cholesterol: activation of HMG-CoA synthase (Hydroxymethyl glutaryl CoA formation step) and HMG-CoA Reductase (mevalonate formation step) Catabolism of Phospholipids The products of these phospholipases are called lysophospholipids and can be substrates for acyl transferases utilizing different acyl-CoA groups. Lysophospholipids can also accept acyl groups from other phospholipids in an exchange reaction catalyzed by lysolecithin:lecithin acyltransferase (LLAT). • phospholipase A2, lysophospholipase, and other enzymes are involved in phospholipid metabolism, • Phospholipase A2 is an important enzyme, its activity is responsible for the release of arachidonic acid from the C-2 position of membrane phospholipids. The released arachidonate is then a substrate for the synthesis of the prostaglandins and leukotrienes. • glycerophosphocholine (GPC) and glycerophosphoethanolamine (GPE) are competitive inhibitors of lysophospholipase activity, inhibits lysophospholipid hydrolysis Anabolism of Phospholipids Choline Acetylcholine CDP-Choline PE - CO2 PS PC + CO2 **Phosphatidylserine (PS) Phosphatidylcholine (PC) CDP: cytidine-5’-diphospho 1,2-diglyceride ** CDP: cytosinediphosphate • DPGs:diphospha tidylglycerols. Also known as cardiolipins are synthesized by the condensation of CDPdiacylglycerol with phosphatidylglyc erols (PG). Clinical Effect • has not yet been fully evaluated, but scientists have studied the role of choline and phospholipids in age related cognitive decline (ARCD), Alzheimer’s disease, and Parkinson’s disease • good dietary intake of phospholipids, cholin lead to an improvement in learning and memory • The fatty acid composition of phospholipids can deteriorate with aging and disease. Regulation of synthesis • The fatty acid distribution at the C-1 and C-2 positions of glycerol within phospholipids is continually in flux, owing to phospholipid degradation and the continuous phospholipid remodeling that occurs while these molecules are in membranes. • Phospholipid degradation results from the action of phospholipases. There are various phospholipases that exhibit substrate specificities for different positions in phospholipids. In many cases the acyl group which was initially transferred to glycerol, by the action of the acyl transferases, is not the same acyl group present in the phospholipid when it resides within a membrane. The remodeling of acyl groups in phospholipids is the result of the action of phospholipase A1 and phospholipase A2 LIPID PROFILE • Is a presentation of concentration of different lipid components in blood. • Normally it involves determination of capillary blood cholesterol and triglyceride of fasting and non-fasting subject. • Concentration of the lipid component is determined using a specific test strips and GCT meter • Low and high readings are indicative to some form of health state. ** refer manual for details Lipid Metabolic Disorder • Abnormalities in the enzymes in lipid metabolism result in 2 types of disorder. 1. Lipidosis : case when there is accumulation of specific fatty substances due to abnormalities in the enzymes that are involved in assimilation of the specific fatty substances eg. Gaucher's Disease, Tay-Sachs Disease, Niemann-Pick Disease Fabry’s Disease; rare case: Wolman's disease, sitosterolemia, Refsum's disease 2. Fatty acid oxidation disorder : When body is unable to properly convert fats into energy due to abnormalities of enzymes in the fatty acid oxidation pathway. Eg medium chain acyl-CoA dehydrogenase (MCAD) deficiency • Gaucher's Disease, most common. - accumulation of glucocerebrosides in liver and spleen, most common in Ashkenazi (Eastern European) Jews leads to enlargment of the organs and brownish pigmentation of skin. - Accumulations of glucocerebrosides in the eyes cause yellow spots called pingueculae to appear in the eye - Accumulations in the bone marrow can cause pain and destroy bone. - 3 types : i) Type 1, the chronic form, with symptom of enlarged liver and spleen and bone abnormalities. More common among adults ii) Type 2, develops in infancy; infants with the disease have enlarged spleen and severe nervous system abnormalities and usually die within a year. iii) Type 3, the juvenile form, can begin at any time during childhood. Children with the disease have an enlarged liver and spleen, bone abnormalities, and slowly develop progressive nervous system abnormalities. Children who survive to adolescence may live for many years. Gaucher's disease can be treated with enzyme replacement therapy • Tay-Sachs disease : accumulate gangliosides in tissues, most common in families of Eastern European Jewish origin. At early age, children with this disease become progressively retarded and appear to have floppy muscle tone. Spasticity develops and is followed by paralysis, dementia, and blindness. Patient usually die by age 3 or 4. Tay-Sachs disease can be identified in the fetus by chorionic villus sampling or amniocentesis. The disease cannot be treated or cured. • Niemann-Pick disease: accumulation of sphingomyelin or cholesterol; has several forms, depending on the severity of the enzyme deficiency and thus accumulation of sphingomyelin or cholesterol. The most severe forms tend to occur in Jewish people. The milder forms occur in all ethnic groups. The most severe form (type A), children fail to grow properly and have multiple neurologic problems. These children usually die by age 3 Type B, disease develops fatty growths in the skin, areas of dark pigmentation, and an enlarged liver, spleen, and lymph nodes; may be mentally retarded. Type C, disease develops symptoms in childhood, with seizures and neurologic deterioration. Some forms of the disease can be diagnosed in the fetus by chorionic villus sampling or amniocentesis. After birth, the diagnosis can be made by a liver biopsy None of the types can be cured.