Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cell-penetrating peptide wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Protein (nutrient) wikipedia , lookup
Gene expression wikipedia , lookup
Western blot wikipedia , lookup
Protein adsorption wikipedia , lookup
Biochemistry wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Protein domain wikipedia , lookup
Protein–protein interaction wikipedia , lookup
List of types of proteins wikipedia , lookup
Proteolysis wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Homology modeling wikipedia , lookup
Pharmacometabolomics wikipedia , lookup
Circular dichroism wikipedia , lookup
Protein structure prediction wikipedia , lookup
Bottromycin wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
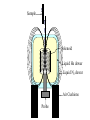
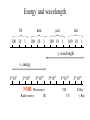
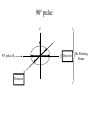
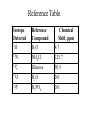
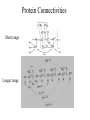
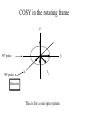
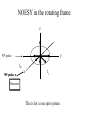
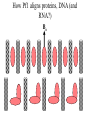
Outline of NMR Spectroscopy Lecture one A. B. NMR Basics I. NMR Phenomenon-Nuclear Spin II. Relaxation III. J coupling IV. Dipolar coupling V. The nuclear Overhauser effect (NOE) VI. Chemical shift Molecular Motion and kinetics I. The NMR timescale II. Exchange III. Molecular motion NMR N uclear M agnetic R esonance NMR takes advantage of an intrinsic dipole moment some nuclei have. Why do some nuclei have an intrinsic dipole moment and other do not? If a nucleus has an uneven distribution of charge, this induces a magnetic field, called spin. This nucleus behaves like a little magnet. In general: if the number of protons or neutrons is odd, then the spin is a multiple of 1/2 (i.e. 1/2, 3/2, etc.). If both are odd, then the spin is 1. If both are even, then the nuclei has no spin-it has an even distribution of charge and is not magnetic. Atoms are little magnets Induced magnetic field Nuclear cloud These little magnets affect each other as a function of DISTANCE to each other. The longest distance that can be “seen” from one atom to another atom is ~5 A. Classic representation of a spin Bo +1/2 The spin precesses in the induced magnetic field, Bo -1/2 Spin motion Bo Spins in field Table of NMR active Nuclei Nuclei Spin # Natural Abundance, % 1H 1/2 99.98 2H 1 0.015 12C 0 98.9 13C 1/2 1.1 14N 1 99.62 15N 1/2 0.4 16O 0 99.76 17O 5/2 0.039 19F 1/2 100 23Na 3/2 100 31P 1/2 100 59Co 7/2 100 What a spectrometer does Sample Solenoid Liquid He dewar Liquid N2 dewar Air Cushions Probe Energy and wavelength M 100 10 mm mm 1 100 10 1 100 10 nm 1 100 10 1 g, wavelength n, energy 3*106 3*108 NMR 3*1010 Microwave Radio waves IR 3*1012 3*1014 VIS UV 3*1016 X-Ray g-Ray Frequency of Nuclei The frequency at which a given nucleus precesses about the induced magnetic field, Bo , is given by the Larmor equation: n= g/2p |Bo| Where g is the gyromagentic ratio for the given nucleus and Bo is in hertz. This is the Larmor frequency (of that nucleus). For example, when someone tells you that they have a 600 MHz NMR instrument, they are telling you the Larmor frequency of the proton nucleus for that magnet. Chemical Shift Chemical Shift is the most basic parameter of NMR and is defined as d in parts per million (ppm). d= (w-w0)/w0 x 106 Where w0 is the Larmor frequency in Hz of the reference line and w is the resonant frequency of the line. The chemical shift of a nucleus originates in the electron cloud around a nucleus, which induces a local magnetic field which opposes the applied field. This leads to the term shielding and deshielding. The more shielded a nucleus is, the larger the chemical shift and conversely, the more deshielded a nucleus is, the smaller the chemical shift. Shielding K. Wuthrich, “NMR of Proteins and Nucleic Acids”. Typical Chemical Shifts Protein chemical shifts DNA and RNA chemical shifts Relaxation Relaxation is the precession of the spin from the perturbed state (x or y) to the relaxed state (z ). There are two ways nuclei do this: 1. Spin-lattice or longitudinal relaxation, T1, which returns magnetization to equilibrium. 2. Spin-spin or transverse relaxation, T2, which leads to loss of phase coherence and signal. 90o pulse z 90o pulse, B1 y Detector x Detector The Rotating Frame T1 z Bo y x Mz = Mo(1-e -t/T1) T1 is the time constant at which the magnetization returns to equilibrium (when all of the magnetization is aligned along z). T1 is experimental determined many ways, but the most common is the inversion recovery method. In structural work, T1 is estimated based on the molecule’s molecular weight (discussed in Molecular motions). 5xT1 is the time for return of >99% of magnetization to equilibrium. This the amount of waiting time between experiments. T2 T2 is the spin-spin relaxation. T2 represents the difference in magnetization between otherwise identical nuclei. T2 has to due with sample inhomogenity and this contributes to line broadening. A simple approximation of T2, T2*: n1/2= 1/pT2*, where n1/2 is the linewidth at half height. z Many different conformations for identical spins, causing slight variations in chemical shift. y x Peak width at half height n n1/2 d, Hz Exchange When a nucleus is exchanging between two or more different environments, this will give rise to complicated spectra. Exchange is important in structural work when you are looking at a dynamic system. For example, if you have a side chain that is moving slowly on the NMR timescale, between two different environments, it can give rise to multiple peaks that can be described by T2. J or scalar coupling Most common coupling between two or more adjacent spin ½ nuclei. The general formula for splitting is: # of lines in the multiplet (N)= 2nI + 1, where I is the spin of the neighboring nuclei and n is the number of identical nuclei. When I=1/2, this simplies to N=n+1. In structure work, J coupling is critical to measuring the bond angles and dihedral angles between nuclei. The Karplus equation is used to calculate the dihedral angles for 3 bond or vicinal coupling: J = A + B cosq+ C cos2q Where A, B, C are coefficients that depend on the nuclei electronegativity. Ethanol – A Classic NMR 1 D experiment H H H C C H H O H TMS d, ppm 0 Dipolar coupling Dipolar coupling is the interaction between two dipoles (of two different nuclei) who are not connected through a bond. This is one of the most important ways nuclei relax. It contributes the most to T1 and is heavily dependent on molecular motion of the molecule. Nuclear Overhauser Effect The NOE is the transfer of magnetization between nuclei. This is the most important effect in structural work. It is important to note that it is heavily dependent on molecular motion (tc) and distance (rIS). A general equation that describes the NOE: R1 = K g2I g2S rIS-6 tc, where I and S are the interacting nuclei. NMR timescale NMR has a broad time scale in terms of what it can “see”. The useful range for structural analysis are from seconds to microseconds. Anything much faster than microseconds, the event tends to be averaged out and a single event is seen. Much slower than seconds leads to line broadening (a T2 effect) and decreasing peaks (NOE effect), which lead to the reduction and sometimes disappearance of the signal. Some useful timescales Average chemical reaction: ns (ps to ms) Average enzymatic reaction: ms (s to ns) Average tumbling time for a globular protein: 107 Hz or 100 ns Average tumbling time for a small molecule: 1011 Hz or 10 ps Molecular motion NMR is very dependent on how molecules rotate in solution. It can be calculated using the Debye equation (assuming a spherical shape): tc= 4pha3/3kT h=viscosity of the solvent and a=radius of the molecule. An approximate tc, assuming typical values for organic solvents, is: tc =10-12 Mw, where Mw is in Daltons. Since this isn’t always the case, relaxation rates (T1 and T2) and NOE measurements can be used to calculate the correlation time. Normally, this is done through other means (like fluorescence anisotropy or DLS). Reference Signal All NMR spectra must be referenced. This means that there has to be a compound that has a known chemical shift in the sample. For example, in organic chemistry NMR, TMS (tetramethylsilane) is used to reference spectra-it is set to zero ppm. For structural work, the reference is set to the most common molecule/frequency. Reference Table Isotope Detected 1H Reference Compound H2O Chemical Shift, ppm 4.7 15N NH4Cl 125.7 13C Glucose 50.5 17O H2O 0.0 31P H3PO4 0.0 Data Processing Raw Data t Fourier transformation Processed data Lecture 2 Outline I. Multidimensional NMR A. B. Why do we need Multidimensional NMR? 2 D NMR 1. Nomenclature 2. Basic experiments a. COSY b. NOESY c. TOCSY/HOHAHA II. C. 3 and 4 D NMR 1. Why we need 3 and 4 D NMR How to make a multidimensional experiment 3. How to read a 3 and 4 D experiment Structural Information and Structure Generation A. Distance Measurements B. Coupling Constants and Angle measurements C. Energy Minimization and Restrained Molecular Dynamics D. Quality of NMR structures d. 3. Multiple Quantum Filtration Solvent supression 2. How to get a protein structure by NMR Clone and express lots of soluble protein Preliminary Spectra (1D 1H, NOESY, COSY) Sequential assignment Collection of conformational contraints Calculation of 3D structure Protein Solutions Not just any protein structure can be solved by NMR If the protein meets these conditions, then it’s a good chance it’s structure can and will be solved by NMR: 1. Mw range: up to 30 kD (for most practical purposes) 2. Soluble at high concentrations (0.5- 1.0 mM) with no aggregation (need to rotate freely) 3. Must be able to purify to 95% or greater 4. Stable at room temperature for days/weeks. Better if relatively temperature insensitive. 5. Must be structured on the NMR timescale Protein Considerations Up to 15-18 kD, it is possible to do the structure without isotopic labeling From 15- ~30 kD, need 13C/15N labeling in the protein From 30 kD and up, not only need 13C, 15N labeling, also need 2H labeling Above 40 kD, need either solid state NMR or special NMR techniques to remove tc requirement in NOE (and other parameters) Isotopic labeling In order for proteins to be isotopically labeled, they must be grown in the presence of isotopically labeled nutrients. This requires some knowledge of metabolism of E. coli or the organism used to produce the labeled protein In the case of E. coli, a strain is used that can grow on minimal media. (i.e. BL21, DH5a) The cells are grown in minimal media made with uniformly labeled 15N ammonium chloride or 13C glucose. Protein purification proceeds as without labeling Why Multidimensional NMR? Multidimensional NMR helps resolve resonances that would otherwise be overlapped and not identifiable. The multidimensional component refers to another frequency (not time, space, etc.) We need to further resolve spectra in biological NMR because our molecules are not simple. Typical Chemical Shifts Protein chemical shifts DNA and RNA chemical shifts 1 D NMR Spectra 1H 1D NMR of Lysozyme 1H 1D NMR of DNA Interesting Facts In a protein/peptide: There are at least 20 spin systems (1 for each amino acid) The average number of protons per amino acid is 8 There are a maximum of ~20 inter-residue contacts per amino acid (in a structured protein/peptide) In DNA/RNA: There are 4-5 spin systems (1 for each base) The average number of protons per base is 11 There are a maximum of ~80 inter-residue contacts per base Experimential design flow chart 2 D NMR- Nomenclature NMR experiments are named in acronyms Some examples: COSY – COrrelation SpectroscopY NOESY- Nuclear Overhauser Effect SpectroscopY TOCSY- TOtal COrrelation SpectroscopY HOHAHA-HOmonuclear HArtman HAhn QF- Quantum Filtered Protein Connectivities Short range Longer range 2 D NMR- Basic Experiments Staple experiments for structural work 1. COSY (1H, 1H) 2. NOESY (1H, 1H) 3. TOCSY/HOHAHA (1H, 1H) These are the basic experiments that form the framework of more specific experiments COrrelation SpectroscopY COSY experiments show who is coupled to who (dipolar coupling and J coupling) This experiment: Confirms the sequence of a protein Sees residues that are coupled together (very close together) This basic experiment can be used in many different ways to look at different couplings (i.e. 1H-13C coupling) and to filter out couplings (quantum filtering) COSY in the rotating frame z 90o pulse 90o pulse, x y x t1 Detector This is for a one spin system COSY-spectrum R N Ca H H O C 1H 1H COSY of Nucleic Acids K. Wuthrich, “NMR of Proteins and Nucleic Acids” COSY of Amino Acids K. Wuthrich, “NMR of Proteins and Nucleic Acids” Quantum Filtering A quantum is a single resonance (also known as Single Quantum, SQ). Double/Triple/Multiple Quantum (DQ, TQ and MQ) are multi-coupled resonances. With filtering, you are selecting for the quantum number so named and is useful when 2 or more step connectivities are required. i.e. DQF-COSY selects for resonances that are coupled twice (hence, Double Quantum). TOtal Correlated SpectroscopY Otherwise known as HOmonuclear HArtman HAhn TOCSY experiments detect indirect J coupling though crosspolarization rather than by relayed magnetization transfer (relayed magnetization transfer experiments are called RELAYCOSY) Cross-polarization (which is caused by spin-locking) causes the spins to become equivalent and give rise to a single resonance. The time it takes for this to happen is equal to 1/2J This experiment is repeated at various spin-lock times in order to measure this coupling constant. The difference between TOCSY and HOHAHA is in how the spin-locking is applied, but these experiments yield the same information TOCSY-spectrum H N Ca H H O C 1H 1H Heteronuclear Correlation Spectroscopy These experiments are also known as HMQC and HSQC (Heteronuclear Multiple/Single Quantum Coherence) This experiment is essentially the same as a COSY, except the magnetization transfer is to another coupled nucleus at another frequency This is done simultaneously with the (usual) proton pulses Homonuclear coupling can interfere with the effect you are trying to observe, so 1H-1H coupling is irradiated (decoupled) The delay in this experiment measures ¼ JIS Heteronuclear couplings are usually very weak effects Nuclear Overhauser Effect SpectroscopY NOESY experiments detect NOEs (magnetization transfer between 2 or more nuclei) This experiment can “see” other nuclei up to 5 A away through space NOESY will give the same information as a COSY, plus the through space information If a standard distance is known and NOEs can be measured, the NOE equation can be simplified to: rIM/rIS = (aIS/aIM)1/6 Where r is the distance between IM and IS and a is the crosspeak intensity Also for this to be true, NOE buildup curves must be done in order to be assured that you are at maximum NOE intensity NOESY in the rotating frame z 90o pulse y tm 90o pulse, x x t1 Detector This is for a one spin system NOEs for DNA Solvent Suppression Very necessary in biological NMR since our samples are in 1H2O. Solvent suppression involves irradiation of the water signal with high power irradiation. This saturates the signal by equalizing the populations of + and – spin states. This can be avoided by putting the sample in 2H2O, but for protein samples, this is not easily accomplished. Pulse sequences 3 D and 4 D NMR Why do we need 3D and 4D experiments? 2D experiments can get very crowded as the number of residues increases 3 and 4D experiments can help resolve spectral overlap by modulating 2 D experiments in various time domains which lets different magnetization buildup (and selecting against other magnetization) 3 and 4 D experiments How to read a 3 or 4 D experiment 2 D to 3 D 2 D to 3 D to 4 D Structural information Experiments are finished, how do we compile the data? How data is fitted Energy Minimization and Restrained Molecular Dynamics This subject is a whole class unto itself. These computational techniques are used to evaluate the model generated by the data, then help correct errors via minimization and dynamics. Modeling is frequently used for sidechains, even residues, that cannot be visualized by NMR (motion is too rapid/slow). Because the data typically gives a range for distances and angles, multiple structures are generated. The more structures generated the better. Quality of NMR Structures Since a lot of structures are generated, one quick quality assessment is the RMSD between them. The RMSD should not be more than 2 A The RSMD equation (for any structure comparison): RMSD = (1/N S (ri – r’i)2)1/2 Where N is the number of atoms being compared, r is the atomic coordinates for the structures in question Another quality assessment for NMR structures is the number of NOEs per residue (both intra/inter residue NOEs) NOEs per Residue A general rule for resolution of NMR structures (to keep nomenclature consistent with Crystallography: Less than 5 NOEs per residue = 8 –10 A structure 6-12 NOEs per residue = 5 A structure 12-15 NOEs per residue = 3 A structure 16-20 NOEs per residue = 2.3-2.5 A structure Over 21 NOEs per residue = 2.0 A structure Clore GM and AM Groenborn (1991) Science Dynamics This is NMR’s real strength. Can tell what atoms are mobile and how fast they vibrate*. In order the evaluate dynamics in NMR, one needs to measure T1, T2 and tc. After these values are obtained, an order parameter is calculated (S2). An order parameter is calculated for all atoms (got the information, why not?) and plotted. In an average molecule there is motion, but mobile regions are visible by large shifts in the order parameter compared to the backbone atoms. Homework Handout Question 1. Nucleus Spin Number Natural Abundance, % 1 1-0 odd 1/2 99.98 2 2-1 even, odd 1 0.015 12 12-6 even, even 0 98.9 13 13-6, odd, even 1/2 1.1 14 14-7, even, odd 1 99.62 15 15-7, odd, odd 1/2 0.4 16 16-8, even, even 0 99.76 17 17-8, odd, even 5/2 0.039 19 19-9, odd, odd 1/2 100 23 23-11, odd, odd 3/2 100 31 31-15, odd, odd 1/2 100 59 59-27, odd, odd 7/2 100 H H C C N N O O F Na P Co Question 2 2A. Without isotopic enrichment, which of the nuclei above would be found in significant levels in biological systems? 1H, 12C, 14N, 16O, 23Na, 31P 2B. How would you enrich a particular protein with an isotope that isn’t at a biologically significant level? To enrich a protein with an isotope, it would need to be produced in the presence of the isotope in question: Cells would need to be grown in the presence of compounds enriched for the isotope(s). Question 3 A. NMR X-ray Solvent conditions more physiological Solvents- uses a lot of detergents, salts Nondestructive Destructive Sees protons, connectivities first, Sees overall physical structure then model physical structure first, have to model in protons, connectivities Can take months, sometimes years Takes minimum 1 month, up to a year Can use as little as 0.1 mM to 1 mM Needs several mM of protein to screen for crystals Can be used to study dynamics Motion is bad Question 3B B. Because of their differences, they can be used to compliment one another, however it has to be certain that the solvent conditions did not change the overall structure of the molecule. Small changes can be accommodated, but large ones will cause contradictions. 1. NMR can confirm that crystallization doesn’t give a structure due to solvent conditions. 2. NMR can help solve the phase problem for crystallography, but 1 has to be true first. 3. If a region appears to be mobile in the crystal, NMR can confirm this and get a minimized structure of the mobile region to complete the picture. 4. If working together, a relatively large protein (25-40 kD) can be solved fast. Problems in the book – 12.2 Consider spin-spin (through bond) splitting of proton peaks for CH3-NH2. A. Diagram the possibilies for the various interactions. B. Show the spectrum with the expected splittings and relative intensities. H H C H N H H ppm Question 12.5 Given that Glu has the following peaks: NH=8.4, aH=4.3, bH=2.1 and 1.9, gH=2.3 for both; at what 2D coordinates do you expect COSY interactions? -OOC HN H H Ca Cb Cg H H H O Cd O- 1H 1H Remember: The knowing the exact chemical shift is not necessary, but knowing the pattern and what each peak represents is! Question 12.6 In the NOESY below, the off-diagonal circles are crosspeaks that were in the corresponding COSY, while the crosses are new crosspeaks in the NOESY. Explain, using proton numbers, what each crosspeak tell us about the molecule. Things to remember: Diagonal are crosspeaks to themselves Peaks that are in the COSY, but not NOESY are correlated to each other New Peaks in the NOESY tell us about non-coupled, spins that are close in space. Question 12.6-NOESY Cell paper “Inhibiting HIV-1 Entry: Discovery of D-Peptide Inhibitors that Target the gp41 Coiled-Coil Pocket Eckert, DM et al. Cell (1999) 99:103-115 What did they do: Discovered a pocket on HIV-1 gp41 and exploited it by designing D-peptides that bind in it. They appear to make good contact and are therefore good inhibitors of HIV-1 infection. They used a variety of structural techniques to confirm binding and contact of the peptides with the pocket. What did they use NMR for? They used NMR to confirm they mode of binding. Specifically, they used NMR to show a shift in the trp571 resonances, which indicate that trp571 is involved in binding of the peptides. This is a good example of the use of shielding! Figures from Cell paper Structural characterization of the complex of the Rev response element RNA with a selected peptide Zhang, Q. et al. Chemistry and Biology (2001) 8: 511-520 What did they do: Did the structure of the RSG-1.2 peptide in complex with the Rev response element RNA because its sequence is different than the natural Rev peptide and therefore its binding should be different than Rev. NMR conditions: Peptide = 19 residues, RNA = 85 bases They used both unlabeled samples and 13C/15N samples They also dissolved their samples into D2O Used 15, 25, 35 and 45 oC for their experiments Figure 1 Figure 2 Figure 3 Figure 4 Figure 5 How good is it? This works out to be: 189 restraints/19 residues = ~10 restraints/residue for the peptide For RNA: 620 restraints / 84 bases = ~7 restraints/base These RMSD measurements are for the converged structures, NOT the structure(s) generated by NMR! Questions to think about 1. Why did they label both the peptide and the RNA? The peptide is small (only 19 residues, Mw ~20Kd)… 2. Why different temperatures? Why would that help? 3. Do you believe this structure? Why or why not? 4. How would you make this better (theoretically, of course)? “Solution structure of cyanovirin-N, a potent HIV-inactivating protein” C.A. Bewley, et al. Nature Structural Biology (1998) 5:571-578 “The Domain-Swapped Dimer of Cyanovirin-N is in a Metastable Folded State: Reconciliation of X-Ray and NMR Structures” L. G. Barrientos, et al. Structure (2002) 10: 673-686 What is cyanovirin-N? A protein isolated from the cyanobacterium, Nostoc ellipsosporum. Identified by a general screening effort to find products with antiviral/HIV activity What does it do? Unknown function in cyanobacterium But…is a very potent inhibitor of HIV How does it work? It prevents fusion of the virus to the host cell (helper T4 cells) by binding to gp120 on the viral surface envelope. Structural Details Cyanovirin-N is an 11 kDa protein (100 residues) Used 15N and 13C labeling to get the structure Used D2O exchange to get some assignments Protein is symmetric-has a 2-fold axis The sample used was 1.4 mM For total restraints per residue: 2509 restraints/100 residues = 25 restraints/residue NOEs per Residue A general rule for resolution of NMR structures (to keep nomenclature consistent with Crystallography: Less than 5 NOEs per residue = 8 –10 A structure 6-12 NOEs per residue = 5 A structure 12-15 NOEs per residue = 3 A structure 16-20 NOEs per residue = 2.3-2.5 A structure Over 21 NOEs per residue = 2.0 A structure Clore GM and AM Groenborn (1991) Science NMR structure of cyanovirin-N This is a collection of over 40 structures generated by “simulated annealing”. Secondary Structure Final 30 structures Parameters Mean structure Figure 6. fig6 Red = negative charge Blue = positive charge Yellow=hydrophobic Comparison of the NMR and Crystal Structures NMR = monomer, X-Ray = domain-swapped dimer In this paper, they reconcile the x-ray structure and NMR structure by generating new ones. They picked conditions to favor dimer formation in NMR (refolding the protein in high concentrations or incubation of the monomer at high concentrations and higher temperature). In the crystal structure, they got a slightly different form, but it (in general) the same as the previous crystal structure. Used the crystal structure as the initial model for solving the solution structure of the dimer. Figure 1 NMR Parameters Looked at the resonance corresponding to Trp49 Ne1H (in b strand 5). This residue serves as part of the interface between the 2 domains- in both the monomer form (A and B – from the monomer) and the domain-swapped dimer form. Used a suspension of Pf1 to help align the protein. Why? R1 = K g2I g2S rIS-6 tc, Figure 7 How Pf1 aligns proteins, DNA (and RNA?) Bo Figure 2 Figure 3 Figure 4 Figure 6 Homework Handout Question 1. Nucleus Spin Number Natural Abundance, % 1 1-0 odd 1/2 99.98 2 2-1 even, odd 1 0.015 12 12-6 even, even 0 98.9 13 13-6, odd, even 1/2 1.1 14 14-7, even, odd 1 99.62 15 15-7, odd, odd 1/2 0.4 16 16-8, even, even 0 99.76 17 17-8, odd, even 5/2 0.039 19 19-9, odd, odd 1/2 100 23 23-11, odd, odd 3/2 100 31 31-15, odd, odd 1/2 100 59 59-27, odd, odd 7/2 100 H H C C N N O O F Na P Co Question 2A A. NMR X-ray Solvent conditions more physiological Solvents- uses a lot of detergents, salts Nondestructive Destructive Sees protons, connectivities first, Sees overall physical structure then model physical structure first, have to model in protons, connectivities Can take months, sometimes years Takes minimum 1 month, up to a year Can use as little as 0.1 mM to 1 mM Needs several mM of protein to screen for crystals Can be used to study dynamics Motion is bad Question 2B B. Because of their differences, they can be used to compliment one another, however it has to be certain that the solvent conditions did not change the overall structure of the molecule. Small changes can be accommodated, but large ones will cause contradictions. 1. NMR can confirm that crystallization doesn’t give a structure due to solvent conditions. 2. NMR can help solve the phase problem for crystallography or crystallography can start as the initial model for NMR, but 1 has to be true first. 3. If a region appears to be mobile in the crystal, NMR can confirm this and get a minimized structure of the mobile region to complete the picture. 4. If working together, a relatively large protein (25-40 kD) can be solved fast. Question 3 In the NOESY below, the off-diagonal circles are crosspeaks that were in the corresponding COSY, while the crosses are new crosspeaks in the NOESY. Explain, using proton numbers, what each crosspeak tell us about the molecule. Things to remember: Diagonal are crosspeaks to themselves Peaks that are in the COSY, but not NOESY are correlated to each other New Peaks in the NOESY tell us about non-coupled, spins that are close in space. Question 3 Figure 8