Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Discovery and development of direct Xa inhibitors wikipedia , lookup
Discovery and development of non-nucleoside reverse-transcriptase inhibitors wikipedia , lookup
DNA-encoded chemical library wikipedia , lookup
Discovery and development of ACE inhibitors wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Discovery and development of cephalosporins wikipedia , lookup
Pharmacognosy wikipedia , lookup
Discovery and development of neuraminidase inhibitors wikipedia , lookup
CCR5 receptor antagonist wikipedia , lookup
Discovery and development of TRPV1 antagonists wikipedia , lookup
Neuropharmacology wikipedia , lookup
5-HT3 antagonist wikipedia , lookup
Discovery and development of antiandrogens wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Drug discovery wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Nicotinic agonist wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Cannabinoid receptor antagonist wikipedia , lookup
Discovery and development of integrase inhibitors wikipedia , lookup
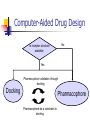
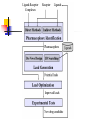
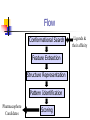
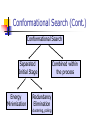
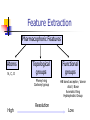
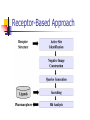
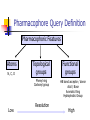
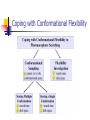
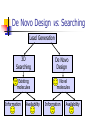
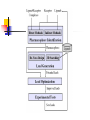
Abstract Drug Design’s goal: to develop new ligands with high binding affinity toward a protein receptor. Pharmacophore: 3D arrangement of essential features that enable a molecule to exert a particular biological effect. Computer-Aided Drug Design No Is receptor structure available Yes Pharmacophore validation through docking Docking Pharmacophore Pharmacophore as a constraint in docking Agenda Definition Computer-Aided Drug Design Flow Pharmacophore Identification Indirect methods Direct methods Pharmacophore fingerprints Applications for Drug Design What is Pharmacophore? Paul Ehrlich (~1900): The molecular framework that carries (phoros) the essential features responsible for a drug's (pharmacon) biological activity. Later: it became clear that the 3D disposition of the pharmacophoric features is also important. Indirect Methods for Pharmacophore Identification Flow Conformational Search Feature Extraction Structure Representation Pattern Identification Pharmacophore Candidates Scoring Ligands & their affinity Input – Dataset of ligands Ligand type Usually active and share the same activity Sometimes inactive ligands are used Rarely different activity levels are used Ligand Diversity Dataset size Usually dataset size < 100 Conformational Search Conformational Search (Cont.) Conformational Search Separated Initial Stage Energy Minimization Redundancy Elimination clustering, poling Combined within the process Feature Extraction Pharmacophoric Features Atoms N, C, O Topological groups Phenyl ring Carbonyl group High Resolution Functional groups HB bond acceptor / donor Acid / Base Aromatic Ring Hydrophobic Group Low Functional Groups Structure Representation The features are combined to form a representation of the whole structure 3 main approaches: 3D point set Graph Set of interpoint distances Pattern Identification Goal definition: MCS (Maximal Common Substructure): find the largest set of 3D features that common to all of the input ligands MCS drawbacks: assume that there is a single common pharmacophore Relaxed MCS: relaxing the requirement that all ligands must possess all features Pattern Identification (Cont.) Relaxed MCS approaches: A small number of ligands may miss a feature of the pharmacophore. A pharmacophore should have at least M features in common with each ligand. Methods: Graph methods (Clique detection) Exhaustive search Genetic Algorithms (GA) Scoring Requirement: the higher the scoring, the less likely it is that the ligands satisfy the pharmacophore model by a chance. The size of a pharmacophore model can sometimes be misleading as a score: 2 charge features > 4 hydrophobes Scoring is more complicated for relaxed MCS Pharmacophore Fingertprints Definition Also termed pharmacophore key 1D descriptor that encodes pharmacophoric information e.g. encodes all the potential n-point pharmacophores that can be present in some conformer of a molecule. Evaluating Molecular Similarity A pharmacophore key for a set of ligands: Union key = the logical OR of the keys of the individual compounds in the set. Molecular Similarity: e.g. Tanimoto coefficient: Tc = NAB/(NA+NB-NAB) 0 ≤ Tc ≤ 1 Pharmacophore Diversity library diversity = #bits set in the union key Designing a diverse library Goal: reduce the number of molecules without decreasing the diversity Process: Diverse Subset Selection Rejecting too rigid or too flexible molecules Calculating pharmacophore fingerprints Rejecting molecules with too few or too many pharmacophores. Iterative selection process Pharmacophore Profiling Direct Methods for Pharmacophore Identication 1. Receptor-Based Approach 2. Complex-Based Approach Negative Image of the Active Site The negative image of the active site is used to construct a pharmacophore model. Receptor-Based Approach Complex-Based Approach Provides information regarding the proteinligand contacts. Important! 1. The active site can be flexible and can rearrange itself to accommodate different ligands. 2. Alternative pharmacophores may be possible within a single binding site. 3. Receptor may have more than one active site. Multiple Alignment for Pharmacophore Investigation. Pharmacophore Applications 1. 2. 3. 4. Pharmacophore Searching. De Novo Design of Ligands. Lead Optimization. 3D-QSAR. Pharmacophore Searching Pharmacophore searching is a part of a more general problem of 3D structure searching. The main aspects in which methods differ from each other: 1. Pharmacophore Query Definition 2. Coping with Conformational Flexibility 3. Pattern Identification Pharmacophore Query Definition Pharmacophoric Features Atoms N, C, O Topological groups Phenyl ring Carbonyl group Low Resolution Functional groups HB bond acceptor / donor Acid / Base Aromatic Ring Hydrophobic Group High Coping with Conformational Flexibility Pattern Identification Graph representation. Solving the sub-graph isomorphism problem by reduction to clique detection. Upper and lower bounds on the interfeature distance constraints. Multi-Level Searching Main concepts: Filter out (ASAP) the compounds that have no chance to satisfy the pharmacophore constraints. The filters are applied in an increasing order of complexity, such that the first are fast and simple while successive ones are more timeconsuming, but are applied only to a small subset. Multi-level Framework 1. Screening Presence of required features/atoms. Comparison of keys/fingerprints. 2. Pattern Identification Substructure searching. 3. Conformational Fitting Only for ‘on the fly’ methods. systematic search, random search, distance geometry, genetic algorithm and directed tweak. Receptor-Based Searching Exploits structural information, like shape and volume. Docking techniques must fulfill: 1. Correct ranking 2. High speed Approaches to Receptor-Based Pharmacophore Searching 1. Pharmacophore-Based Prescreening Prescreens the database using the pharmacophore information and then docks the selected candidates. 2. Pharmacophore-Constrained Docking Incorporates pharmacophore information into the docking process. Define the target binding site points. Match the distances. Calculate the transformation matrix for the orientation. Dock and score the molecule. Pharmacophore-Constrained Docking: FlexX-Pharm 1. Ligand fragmentation 2. Select & Place a set of base fragments 3. Construct the ligand by linking the remaining fragments. New: A set of predefined pharmacophore requirements must be fulfilled! De Novo Design of Ligands A pharmacophore model can be used in De novo design to construct novel ligands that satisfy the physicochemical constrains. De Novo Design vs. Searching Lead Generation 3D Searching Existing molecules Information Availability De Novo Design Novel molecules Information Availability Use of Pharmacophores in Lead Optimization Lead optimization is the process in which a biologically active compound is modified to fulfill all the properties that are required from a drug (e.g. physicochemical and ADME/Tox properties). Pharmacophore can direct chemists to include/exclude specific chemical groups. QSAR Quantitative Structure Activity Relationships (QSAR) uses statistical correlation methods, to predict quantities such as: binding affinity the toxicity the pharmacokinetic parameters. Use of Pharmacophore in 3D-QSAR 3D-QSAR analyses the correlation between the structural features and the biological activity. Pharmacophore models can be used to generate good alignments suitable for QSAR analysis. Summary Plays a key role in CADD, especially in the absence of receptor structure. Can suggest a diverse set of compounds with different scaffolds. Important! 1. Necessary but insufficient condition. 2. Several binding modes and several pharmacophores within the same active site. 3. Several active sites. Pharmacophore-Based Prescreening Approach Use the receptor active site to derive a pharmacophore query. Search the DB of candidate ligands. Dock the ligands into the receptor active site and score.