Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Neuropsychopharmacology wikipedia , lookup
Discovery and development of tubulin inhibitors wikipedia , lookup
Discovery and development of ACE inhibitors wikipedia , lookup
Discovery and development of direct Xa inhibitors wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Discovery and development of direct thrombin inhibitors wikipedia , lookup
Neuropharmacology wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
Discovery and development of cephalosporins wikipedia , lookup
Pharmacognosy wikipedia , lookup
Drug interaction wikipedia , lookup
Discovery and development of neuraminidase inhibitors wikipedia , lookup
Drug design wikipedia , lookup
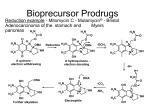
Prodrug strategies to overcome poor water solubility and permeability Pharmaceutical Technology 1 Prodrugs A prodrug is a poorly active or inactive compound containing the parental drug that undergoes some in vivo biotransformation through chemical or enzymatic cleavage, enabling the delivery of the active molecule at efficacious levels. Carrier-linked prodrugs can be classified as bipartite prodrugs, in which the carrier is linked directly to the parent drug, and tripartite prodrugs, in which a spacer links the carrier to the parent drug . Carriers are commonly attached by chemical groups such as ester, amide, carbamate, carbonate, ether, imine, phosphate, among others Mutual prodrugs are a type of carrier-linked prodrug in which two active compounds are linked each acting as the carrier to the other. These prodrugs have increased effectiveness Through synergistic action. Another type of carrier-linked prodrug is the macromolecular prodrug; these prodrugs use polymeric backbones as carriers. 2 3 Prodrugs • Prodrugs also have been synonymously referred to as latentiated drugs, bioreversible derivatives, and congeners. • However, the term prodrug gained wider acceptance and usually describes compounds that undergo chemical transformation within the body prior to exhibiting pharmacologic activity. Some of the earliest examples of prodrugs are methenamine (antibiotic, UTI) and aspirin. 4 In the late nineteenth century a chemist, Felix Hoffman in Bayar Company, synthesized the antipyretic agent Aspirin (acetylsalicylic acid), which was introduced for the first time in clinical practice in 1899; it can be considered a less corrosive prodrug form of salicylic acid to minimize the gastric irritation and ulcerogenicity associated with salicylic acid. Methenamine was discovered in 1899 by Schering as inactive prodrug that delivers the antibacterial formaldehyde. It is useful in the treatment of urinary tract infection, when transported to urinary bladder it becomes acidified to provide a medium in which formaldehyde is generated. 5 Rationale for Prodrug Design • A large number of the new molecular entities with promising therapeutic profiles are dropped from the screening stage because of their inferior physicochemical and biopharmaceutical properties. • These undesired properties result in poor absorption, extensive metabolism, and low bioavailability because of physical, biological, or metabolic barriers. • If the chemical structure of the drug or lead compound can be modified to overcome these barriers and then revert to the pharmacologically active form, the drug can 6 be delivered efficiently. Rationale for Prodrug Design • The objective of designing of prodrugs is to achieve: – Favorable physicochemical characteristics (e.g., chemical stability, solubility, taste, or odor) – Favorable biopharmaceutical properties (e.g., oral absorption, first-pass metabolism, permeability across biological membranes such as the blood-brain barrier, or reduced toxicity) – Favorable pharmacodynamic properties (e.g., reduced pain or irritation). 7 Enhancement of solubility/dissolution through prodrug formation • To solve the problem of poor solubility/dissolution of a drug through prodrug formation we must understand the reason why is the compound exhibiting poor solubility: – Is the molecule one that displays high crystallinity, a “brick dust” molecule, or – is it a low melting or “grease ball” molecule? • Obviously, a continuum exists, with most molecules not fitting either extreme. 8 Enhancement of solubility/dissolution through prodrug formation • If the molecule has the characteristics of a “grease ball” molecule and usual formulation approaches do not work, solubility enhancement through the use of a polar promoiety may prove useful. • If one has a “brick dust” molecule a polar promoiety may work, as might strategies that disrupt the intermolecular interactions that led to the high crystallinity 9 Addition of polar functionalities • Conventional wisdom dictates that placing a polar functional group in the structure of a molecule with limited aqueous solubility should enhance solubility. • In the case of a prodrug, that functionality would have to be removed/modified, either chemically or enzymatically, to regenerate the parent drug. 10 Addition of polar functionalities • Many prodrugs designed to increase water solubility involve the addition of an ionizable promoiety to the parent molecule. • Because charged molecules have greater difficulty crossing biological membranes, one must balance increased water solubility with the potential for decreased permeability. 11 Addition of polar functionalities • One might argue that a phosphate ester of a drug with an alcohol functionality in its structure would produce a poorly, membrane permeable prodrug. • However, phosphate esters have been shown to be very effective at improving the delivery of poorly water-soluble parent drug molecules after oral delivery. 12 Addition of polar functionalities • For R–OH, a highly permeable drug, bioavailability after oral dosing is limited by slow dissolution. • The prodrug, R–OPO3= is much more soluble, rapidly dissolves in the content of the GIT but is cleaved to R– OH by the presence of the enzyme, alkaline phosphatase, seen in abundance on the brush border surface of the cells lining the small intestine, the enterocytes. • R–OH, being permeable, readily crosses the enterocyte membranes and enters the systemic circulation. 13 Addition of polar functionalities • limitations of this approach: – The phosphate prodrug must be a good substrate for alkaline phosphatase – R–OH must be permeable once cleaved – Too rapid a cleavage of a very insoluble R–OH can result in precipitation of R–OH, and thus poor redissolution 14 Addition of polar functionalities • Fosamprenavir, a phosphate prodrug of the HIV protease inhibitor, amprenavir. • Amprenavir was originally formulated in a 150 mg capsule containing TPGS (d-alpha tocopheryl polyethylene glycol 1000 succinate ), PEG 400 and propylene glycol, requiring patients to take 8 capsules to achieve a dose of 1200 mg twice a day. d-αTocopheryl polyethylene glycol 1000 succinate (simply TPGS or Vitamin E TPGS) is formed by the esterification of Vitamin E succinate with polyethylene glycol 1000. As novel Generally Regarded as Safe (GRA) nonionic surfactant, it exhibits amphipathic properties and can form stable micelles in aqueous vehicles at acid 0.02 wt%. concentration asSuccinic low as Tochopherol PEG •15 This puts amprenavir at a competitive disadvantage to other lower dose and more conveniently administered antiAIDS drugs. Addition of polar functionalities • Amprenavir has a secondary alcohol group in its structure that was synthetically phosphorylated to produce fosamprenavir. • The commercial success of fosamprenavir has made an impact. • Fosamprenavir is in the form of a calcium salt which is approximately 10 times more soluble than amprenavir. 16 Addition of polar functionalities • Because of this superior solubility, even more so at low pH values where the calcium salt dissociates, fosamprenavir can be formulated as a 700 mg tablet (equivalent to 600 mg of amprenavir) thus reducing the dosing to 2 tablets twice a day. • The oral availability of amprenavir from fosamprenavir is essentially equivalent to amprenavir from the original capsules. 17 A similar approach was not successful for propofol, a phenolic compound used as a systemic anesthetic by injection or infusion. The phosphat prodrug was too stable in blood after injection. Therefore, a (phosphonooxy)methyl ester prodrug (fospropofol) was synthesized (see Scheme 3), the disodium salt of which is >1000-fold more soluble than the parent drug. The (phosphonooxy)methyl prodrug is readily cleaved in vivo by phosphatases, followed by chemical hydrolysis yielding propofol, phosphate, and formaldehyde. 18 Disulfide prodrugs A paclitaxel prodrug containing a disulfide moiety was described as an anticancer compound. The rationale behind the design of these molecules is based on the recognition of disulfide subunit by the enzyme glutathione, which releases the paclitaxel. High levels of this enzyme have been described in cancer cells, and one hypothesis speculates that this enzyme is involved in resistance to many anticancer drugs. The prodrug compounds were 6–100 times more soluble than paclitaxel. Moreover, the most active compound 18 was 65 times more water soluble than paclitaxel. For this molecule, superior antitumoral activity was identified in all types of cells evaluated, except for MCF10A. After oral administration, the bioavailability in mice was five-fold greater than that of the parental drug. 19 an intracellular sulfhydryl-containing species such as glutathione (GSH or its thiolate anion GS- at biological pH) would attack the disulfide bond in the prodrug to give the transient metabolites [M1] and [M2]. The thiolate anion in [M1] would trigger intra-molecular cyclization by attacking the nearby carbonate group to release paclitaxel (1) and the cyclic ethylene monothiolcarbonate [M3].The metabolite [M2] could be excreted or metabolized further by plasma esterases if it isbonded to a water-soluble group or moiety via an ester bond. 20 Macromolecular prodrug Oridonin is a natural product is found in Rabdosia rubescens, a Chinese medicinal plant, and it exhibited antitumoral activity against several types of cancer, including leukemia. Exploring polyethylene glycol as carrier, the authors linked oridonin to PEG, using succinic acid as a spacer. Four different molecular weights of PEGs were tested to increase solubility in water. The greatest increase in solubility was observed for a low PEG-molecular-weight (5 kDa) conjugate 10. This prodrug had 99.2 times the solubility of oridonin. A chemical hydrolysis study showed a sustained-release effect for the prodrugs. In vivo studies using the two intermediate conjugates (10 and 20 kDa) demonstrated a successful use of this prodrug approach, as these derivatives have better pharmacokinetic profiles than oridonin 21 Decreased crystal packing • A strategy not often considered to effect better oral delivery of poorly soluble drugs is one that attempts to convert a “brick dust” molecule to a “grease ball” molecule. • Noyes–Whitney model describes the dissolution rate (DR) of a drug under sink conditions, 22 Decreased crystal packing • Although this equation has some limitations, it illustrates that DR should be proportional to solubility, Cs, but solubility in what? • Because of the presence of mixed micelles of bile salts and lecithin as well as food digestion products, the GIT presents an environment favorable to dissolving the poorly soluble, lipophilic compouds. 23 Decreased crystal packing • The N–H at the 3-position of phenytoin is known to hydrogen bond with the carbonyl of a second phenytoin molecule. Thus, removing or blocking the N–H group at the 3-position dramatically changes the properties of the molecule. • The melting points of a series of 3-acyloxymethyl prodrugs of phenytoin is shown along with their solubility in water, cyclohexane and in a bile salt/lecithin mixture used to simulate the GIT contents (referred to by the acronym SIBLM, for simulated bile salt, lecithin mixture). 24 Decreased crystal packing 25 Decreased crystal packing • Note the melting behavior of the pentanoyl- (C4H8CO-) and octanoyl- (C7H15CO-) derivatives, which have melting points lower than their higher and lower homologs, and the relationship between melting point and the solubility of the various prodrugs in the hydrocarbon solvent, cyclohexane. • Clearly if one were to only consider water solubility and DR in water, phenytoin itself would be the superior candidate. When one considers the DR in the SIBLM however, the choice becomes less clear, with the DR of the octanoate prodrug superior to that of phenytoin in this medium 26 Decreased crystal packing • The oral, absolute bioavailability of phenytoin from phenytoin, the pentanoate and the octanoate was assessed in fed and fasted dogs. 27 Decreased crystal packing • The bioavailability of phenytoin from the two prodrugs is superior in both the fed and fasted state despite their lower aqueous solubility. In the fed state, phenytoin bioavailability from the pentanoate and the octanoate is close to complete, even though the octanoate had limited aqueous solubility and no measurable DR in water. 28 Mutual prodrugs • This azo bond is stable in the upper GIT and is cleaved in the colon by the azoreductases produced by the microflora • Sulphasalazine was introduced for the treatment of rheumatoid arthritis and anti-inflammatory disease. Chemically it is salicylazosulphapyridine (SASP), where sulfapyridine is linked to a salicylate radical by an azo bond (mesalazine). When taken orally, only a small proportion of the ingested dose is absorbed from the small intestine and the bulk of the sulphasalazine reaches the colon intact. There it is split at the azo bond by the colonic bacteria with the liberation of sulphapyridine (SP) and 5-ASA 29 Enhancing Permeability and Absorption The transport of a drug to its site of action usually requires passage through several lipid membranes; therefore, membrane permeability has a considerable influence on drug efficacy. In oral drug delivery in particular, which is the preferred route for the majority of drugs, the most common absorption routes are unfacilitated and largely nonspecific, passive transport mechanisms. The lipophilicity of poorly permeable drugs can be increased by modifying the hydrocarbon moieties. However, good activity sometimes requires a structure, which is far from ideal one for good membrane permeability. In such situation, the prodrug strategy can be an extremely valuable option. Improvements of lipophilicity have been the most widely studied and therefore now also the most successful field of prodrug research. It has been achieved by masking polar ionized or nonionized functional groups to enhance Oral drug absorption. A hydrophilic hydroxyl, thiol, carboxyl, phosphate, or an amine group on the parent drug can be converted to more lipophilic alkyl or aryl esters, and these prodrugs are readily bioconverted to their active species by ubiquitous esterases, which are present throughout the body. The attractiveness of this prodrug approach is that the alkyl chain length can be modified to obtain precisely the desired lipophilicity. Currently, a considerable number of ester prodrugs have advanced into clinical use. 30 HA.PT Oseltamivir (Tamiflu; Hoffmann-La Roche Inc., Nutley, NJ) is an orally active ethyl ester prodrug of a selective inhibitor of viral neuraminidase glycoprotein and used in the treatment of influenza types A and B . After absorption, oseltamivir undergoes rapid bioconversion to its parent drug mostly by the action of carboxylesterase The bioavailability of the more lipophilic oseltamivir is almost 80%, whereas the corresponding value for free carboxylate is as low as 5%. 31 HA.PT Adefovir dipivoxil (Hepsera; Gilead Sciences, Inc., Foster City, CA) represents a more recent example of a prodrug, in which the ester promoiety is attached to a phosphoryl group on the parent drug via a spacer. This pivaloyloxymethyl phosphoric acid ester of the nucleotide reverse transcriptase inhibitor adefovir (PMEA) is given orally to treat retro-, herpes-, and hepadnaviruses and has almost four times greater bioavailability than adefovir itself. 32 HA.PT The latest clinical example of a more lipophilic oral prodrug is dabigatran etexilate (Pradaxa; Boehringer Ingelheim Pharma GmbH, Ingelheim, Germany), a direct thrombin inhibitor for stroke prevention, which has been available in Europe and many other countries since 2008. However, it was not until the October 2010 when dabigatran etexilate became the first oral alternative to warfarin to be approved in the United States. The active drug dabigatran is a very polar, permanently charged molecule with a log P of -2.4 (noctanol/buffer, pH 7.4) and therefore it has zero bioavailability after oral administration. In the more lipophilic bifunctional prodrug dabigatran etexilate, the two polar groups, the amidinium moiety and carboxylate, are masked by carbamic acid ester and carboxylic acid ester groups, respectively, which results in better absorption with bioavailability of 7% after oral administration 33 HA.PT Another method to increase oral absorbtion is to design prodrugs, which have structural features similar to substrates that are absorbed by carrier-mediated transport. This strategy is particularly important when passive transcellular absorption is negligible. Good examples of carrier-mediated of prodrugs are midodrine (ProAmatine, Gutron; Shire Llc, Florence, KY) and valacyclovir (Valtrex; SmithKline Beecham, Philadelphia, PA; Glaxo Wellcome Inc., Research Triangle Park, NC). Midodrine is a glycine prodrug of desglymidodrine (DMAE), a selective a1-receptor agonist used in the treatment of orthostatic hypotension, in which the glycine promoiety is attached to an amine group of DMAE. Midodrine is absorbed via the intestinal proton-coupled peptide transporter 1 and is bioconverted by asyet-unknown peptidases, mainly in the liver and systemic circulation It has an oral bioavailability of 93%, which is significantly more than the corresponding value for DMAE (50%). 34 HA.PT Valacyclovir is a L-valyl ester prodrug of acyclovir, a purine nucleoside used for the treatment of herpes virus infections. Valacyclovir is a substrate, not only for peptide transporter 1 but also for Na+-dependent neutral amino acid transporter. After absorption, it is bioactivated by valacyclovir hydrolase.The bioavailability of this prodrug is more than 50%, which is 20 to 35% better than the bioavailability of acyclovir. Valacyclovir is actually apreprodrug or double prodrug, because after the hydrolysis of the valine promoiety, acyclovir, like all nucleosides, requires phosphorylation before it forms the active nucleotide triphosphate. The first phosphorylation reaction is mediated by viral thymidine kinase, which is far more effective than the endogenous enzyme in uninfected cells. This further improves the selectivity of acyclovir. Cellular kinases subsequently phosphorylate the nucleotide monophosphate to its active triphosphate form, which then can act as a selective inhibitor of viral infection. 35 HA.PT 36 HA.PT