Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Immune system wikipedia , lookup
Lymphopoiesis wikipedia , lookup
Adaptive immune system wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Hygiene hypothesis wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Innate immune system wikipedia , lookup
Sjögren syndrome wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
Pathophysiology of multiple sclerosis wikipedia , lookup
Autoimmunity wikipedia , lookup
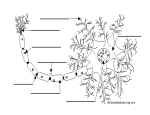
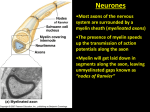
© 2001 Nature Publishing Group http://immunol.nature.com N EWS & V IEWS Multiple sclerosis: a two-stage disease The pathogenesis of multiple sclerosis consists of an inflammatory and neurodegnerative phase. Better understanding of these stages has aided the development of specific therapeutic targets. © 2001 Nature Publishing Group http://immunol.nature.com LAWRENCE STEINMAN Multiple sclerosis (MS) usually begins in early adulthood with an autoimmune inflammatory “strike” against components of the myelin sheath. Paralysis, sensory disturbances, lack of coordination and visual impairment are common features. The disease often starts with an “attack” that lasts from a few days to weeks; this is followed by remission that lasts from a few months to years. This relapsing-remitting phase often lasts five to ten years, but ∼30% of individuals with this form of MS enter a secondary chronic-progressive state. This chronicprogressive state is often characterized by the inability to walk, which leaves the MS patient wheelchair-bound. In the chronic-progressive phase, distinct attacks are rare and the disease progresses insidiously. Occasionally, however, clinical disability begins with this progressive phase, in which case the disease is called “primary-progressive MS”. Evidence indicates that the earlier phase of disease, characterized by distinct attacks followed by remission, may be mediated by an autoimmune reaction. The subsequent chronic phase of disease is due to degeneration of both the myelin sheath, which is synthesized by oligodendroglial cells, and the underlying axon, which emanates from the neuronal cell body some distance away. Indeed it is axon loss in the spinal cord and spinal cord atrophy that correlate most strongly with the inability to walk and paralysis1,2. Worldwide, approximately 1,000,000 individuals are afflicted with MS. Women with the disease outnumber men two to one. This bias towards females is seen in other autoimmune diseases, for example, rheumatoid arthritis, systemic lupus erythematosus and thyroiditis. Genome-wide studies have revealed that susceptibility to MS is linked to genes in the major histocompatibility complex (MHC) on chromosome 63–5. Alleles for certain class II genes, HLA-DR and HLA-DQ, confer the strongest risk of contracting MS. Other genes within the HLA complex are involved in the pathogenesis of MS, including expression of tumor necrosis factor-α (TNF-α), various components of the complement cascade and myelin oligodendroglial glycoprotein. More recently, transcriptional profiling with gene microarrays and largescale sequencing of transcripts from MS lesions have identified a number of genes that are involved in the pathogenesis of acute disease. 762 These include immunoglobulin and interleukin 6 (IL-6) as well as osteopontin, which plays a role in the transition from relapsing-remitting to chronic MS (Fig. 1a). We have recently learned, from sequencing the human genome and the genomes of various microbes, that biological organisms share many genes. Hence various proteins are used, in a modular manner, to build structures whose intrinsic components can resemble each other. If a human and a microbe invading that human share a common gene sequence that encodes one of these conserved structural motifs, the immune system, in recognizing a structure on this foreign microbe, may mistakenly also attack “self”. In the context of MS, many microbial protein sequences share homologies with structures found on the myelin sheath; this leads to an attack on myelin via a process called molecular mimicry. Relapses in MS are often triggered by common viral infections. Viruses such as herpesvirus 6, influenza, measles, papilloma virus and Epstein-Barr Virus all have genes encoding sequences that mimic those found in the major structural proteins of myelin. Indeed, antibodies to components of the myelin sheath cross-react and bind sequences from these microbes. T cells also recognize sequences from the myelin sheath that are shared with these microbial sequences6. Once a T cell, B cell or macrophage is activated by a foreign microbe, self-protein or microbial superantigen, it may penetrate the blood-brain barrier. Penetration of the blood-brain barrier by activated lymphocytes is a multistep process (Fig. 1). There are specialized capillary endothelial cells in the central nervous system (CNS) that are nonfenestrated and connected through tight junctions. During the inflammatory response, TNF-α and interferon-γ (IFN-γ) induce these capillary endothelial cells to express vascular cell adhesion molecule (VCAM) and MHC class II molecules. Activated T cells express integrins, such as very late antigen (VLA-4), and members of the immunoglobulin superfamily, such as CD4, that can bind VCAM and MHC class II molecules, respectively. Once activated, any T cell expressing VLA-4, for example, can bind to adhesion molecules on the surface of inflamed endothelium and “walkthrough” the endothelium. In an animal model nature immunology • volume 2 no 9 • september 2001 • of MS, acute experimental autoimmune encephalomyelitis (EAE), blockade of VLA-4 reverses clinical paralysis and prevents further relapses in the chronic model of this disease. In acute MS lesions, VLA-4 is found on T cells that collect in the “perivascular lymphocyte cuff”, a region around veins and capillaries that is limited by the extracellular matrix. Clinical studies with a human antibody to VLA-4 are now in Phase III following promising Phase II trial results in which the incidence of MS relapses was reduced. Once the activated lymphocytes have extravasated, they still must pass through a barrier of extracellular matrix, comprised of type IV collagen, before they can enter the CNS. Matrix metalloproteases (MMPs) are a family of structurally and functionally related enzymes that are involved in the degradation of the extracellular matrix as well as the proteolysis of myelin components in MS. MMPs contain Zn2+at their active site, show TNF-α convertase activity and induce the cleavage of TNF-α from a cell-bound to a soluble form. Gelatinase A and B (also called MMP2 and MMP9) play a key role in penetration of the extracellular matrix. These MMPs are detectable in the spinal fluid of MS patients, and gelatinase B immunoreactivity is present in endothelial cells, pericytes, macrophages and astrocytes of MS lesions7. Myelin-specific T cell clones derived from MS patients also produce gelatinase B upon activation with antigen. The presence of gelatinase B in the perivascular infiltrate is associated with disruption of the type IV collagen-positive basement membrane and is critical in the opening of the blood-brain barrier. Once the blood-brain barrier is breached, inflammatory cells spread into the white matter of the CNS. MMP inhibition by tissue inhibitors of matrix metalloproteases (TIMPs) can block TNF-α and thereby downregulate the induction of adhesion molecules such as VCAM. TIMP-1 is present in the spinal fluid of MS patients and is inducible by various cytokines, including TNF-α. In terms of MS therapy, IFN-β, a potent inhibitor of gelatinase B activity, has been used relatively successfully in clinical trials. Inhibition of gelatinase is thought to interfere with T cell migration into the CNS as well as T cell secretion of TNF-α. Other MMP inhibitors are currently under http://immunol.nature.com © 2001 Nature Publishing Group http://immunol.nature.com N EWS & V IEWS a Inflammatory phase Antigen-presenting cell Neuron VCAM T cell receptor VLA-4 Myelin fragment Complement Myelin sheath T cell reentry into circulation Axon B cell Venule Collagen type IV on the ECM Adhesion and penetration of blood-brain barrier Naked axon Antibody Cytokines and chemokines eg TNF-α, IFN-γ, IL-12, Osteopontin and IL-6 Macrophage Demyelination Release of toxic mediator b Degenerative phase Autoreactive T cell PLP brain-specific isoform Excess glutamate Glutamate receptor AMPA or kainate receptor MHC class II Antigenpresenting cell Myelin sheath Axon Necrosis Glutamate receptor AMPA or kainate receptor Terminal axon bulb Bob Crimi © 2001 Nature Publishing Group http://immunol.nature.com MHC class II Figure 1.The two stages of the progression of MS. (a) Autoimmune attack. (b) Neurodegeneration. (ECM, extracellular matrix) intense development for clinical MS trials. Once immune cells have spread to the white matter of the CNS, the immune response is targeted to the entire supramolecular myelin complex. Antibodies to various myelin proteins and lipids of the myelin sheath, as well as to molecules expressed in the CNS, are secreted by B cells that have migrated to the brain or from serum that has extravasated across the bloodbrain barrier6,7. Activated complement proteins appear in the spinal fluid along with membrane-attack complexes, which represent the terminal components of this cascade. T cells target certain proteins normally found in the myelin sheath. These include myelin basic protein, myelin oligodendroglial glycoprotein and http://immunol.nature.com • september 2001 • volume 2 no 9 • proteolipid protein, as well as stress proteins such as αB crystallin, which is found in the myelin sheath after activation via the inflammatory response. The T cells produce cytokines, notably lymphotoxin-α (LT-α) and TNF-α, which are members of the TNF family. LT-α is secreted as a LT-α3 homotrimer and, like TNF-α, can bind to the p55 TNF receptor (p55-TNFRI) or the p75 TNFR (p75-TNFRII). These cytokines induce macrophages, microglial cells and astrocytes to produce NO and osteopontin. The free radical NO is a major mediator in autoimmune diseases. NO is involved in the killing of oligodendroglial cells by microglia. Nitric oxide synthase (iNOS), which catalyzes NO synthesis, has been found in demyelinating lesions in MS. Both IFN-γ and TNF-α induce iNOS transcription in astrocytes, microglia and macrophages. The combined effect of antibody, complement, NO and TNF-α damages myelin and induces the macrophage to phagocytose large chunks of the myelin sheath. In addition, macrophages and T cells produce osteopontin. This induces more T helper subset 1 (TH1) cytokines, including IFN-γ and IL-12, and down-regulates TH2 cytokines such as IL10. TH1 cytokines may exacerbate MS, whereas TH2 cytokines may reduce the extent of MS lesions8. This concerted attack by T cells, B cells, complement and inflammatory mediators such as cytokines, osteopontin and NO produces areas of demyelination, which impairs electrical conduction along the axon and produces the pathophysiological defect. TNF-α, LT and other members of the TNFR family may also play key roles in the pathogenesis of oligodendroglial damage. TNF expression is elevated in the spinal fluid in MS relapses; TNF has also been found in MS lesions. In addition, myelin basic protein–reactive T cells from HLA-DRB1*15-positive MS patients express increased amounts of TNF-α. Experimental trials with altered peptide analogs of myelin basic protein that down-regulate the expression of TNF and up-regulate TH2 cytokines can decrease the size of new lesions in white matter8. Similarly the approved drug, Copaxone, induces a shift towards TH2 cytokine production by myelin-reactive T cells. This reduces the frequency of relapses in early MS and decreases the degree of inflammatory activity in white matter. During chronic MS, when exacerbations and remissions of MS are rare, there is evidence for axon loss and atrophy of the brain and spinal cord1,2,9. In EAE and in MS, AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazoleppropionic acid), which mediates toxicity induced by the excitatory neurotransmitter nature immunology 763 © 2001 Nature Publishing Group http://immunol.nature.com © 2001 Nature Publishing Group http://immunol.nature.com N EWS & V IEWS gluatamate, is present on oligodendroglial cells and neurons. During inflammation in both MS and EAE, lymphocytes, brain microglia and macrophages release excessive amounts of glutamate, which then activate AMPA receptors (Fig. 1). Blockade of these receptors with antagonists can ameliorate EAE, including clinical relapses when treatment is begun after the onset of paralysis10. The blockade of AMPA receptors does not influence the immune response to myelin antigens but somehow protects oligodendroglial cells and axons from immune-mediated damage. Damage may be due to increased fluxes of calcium, which may cause necrotic damage to oligodendroglial cells and axons. The use of neuroprotective agents that block glutamate receptor subtypes is the key focus in the development of new therapies for stroke and neurodegenerative conditions, and this approach could prove useful for treatment of the chronic degenerative phase of MS as well. The recognition of an inflammatory and a neurodegenerative phase of MS has facilitated the targeting of therapies that are specific for various stages of MS. Thus, drugs such as IFN-β interfere with lymphocyte migration to brain; altered peptides and Copaxone affect cytokine production by autoimmune T cells; and glutamate receptor antagonists block the Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity insidious atrophy and death of oligodendroglial cells and the underlying axon. 1. 2. 3. 4. 5. 6. 7. Trapp, B. D. et al. New Engl. J. Med. 338, 278–285 (1998). Loseff, N. A. et al. Brain 119, 701–708 (1996). Haines, J. L. et al. Nature Genet. 13, 469–471 (1996). Ebers, G. C. et al. Nature Genet. 13, 472–476 (1996). Sawcer, S. et al. Nature Genet. 13, 464–468 (1996). Wucherpfennig, K.W. et al. J. Clin. Invest. 100, 1114–1122 (1997). Conlon, P., Oksenberg, J. R., Zhang, J. & Steinman, L. Neurobiol. Dis. 6, 149–166 (1999). 8. Kappos, L. et al. Nature Med. 6, 1176–1182 (2000). 9. Lucchinetti, C. F., Bruck,W., Rodriguez, M. & Lassmann, H. Brain Pathol. 6, 259–274 (1996). 10. Pitt, D.,Werner, P. & Raine, C. Nature Med. 6, 67–70 (2000). Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, CA 94305, USA. ([email protected]) B cells can regulate many aspects of immune reactivity, as well as differentiate into antibody-producing cells. In SLE, a systemic autoimmune disease, recent research suggests enhanced B cell function is the defining pathogenic event. PETER E. LIPSKY Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterized by the production of numerous autoantibodies and involvement of skin, joints, kidneys, brain, serosal surfaces, blood vessels, blood cells, lungs and heart. As opposed to lupus in animal models, SLE in humans is heterogeneous and affects different individuals with a wide range of disease courses and manifestations. In addition, the progression of SLE in an individual is difficult to predict at the onset, although a number of factors are associated with more frequent or aggressive disease. These include female gender, AfricanAmerican and African-Caribbean origin and restricted educational experience1. SLE has been considered to be the prototypic systemic autoimmune disease because it is associated with the production of a host of autoantibodies, some of which appear to have pathogenic consequences1,2. In general, systemic autoimmune diseases have been distinguished from organ-specific autoimmune diseases. The latter are thought to develop as a result of immune responses to a limited set of specific autoantigens or cross-reactive exogenous antigens, whereas the former are believed to emerge from a more global abnor764 mality in immunoregulation. Indeed, for most autoimmune conditions, it remains uncertain whether such a clear distinction can be made because systemic abnormalities, such as defects in pathways of apoptosis, may lead to limited expression of autoimmunity in humans. Implicit in this delineation is the distinction between autoimmunity and autoimmune disease. Autoimmunity, the production of autoantibodies or the activation and expansion of T cells that react with autologous cells, proteins or tissues, is relatively common. Autoimmune disease, in which selfreactivity leads to tissue pathology, is much less common. In most circumstances, the determinants of tissue damage and their relationship to autoimmunity have not been fully determined. SLE is characterized by diffuse autoimmunity and characteristic tissue pathology. Despite the presence of both autoantibodies and tissue pathology in SLE, the relationship remains controversial and exact explanations for many of the clinical manifestations of this disease remain unknown. Genetically determined characteristics of the immune response—including its specificity, intensity or resolution as well as the quality, magnitude nature immunology • volume 2 no 9 • september 2001 • or diversity of the effector molecules produced or the nature of the end organs themselves—may contribute to tissue pathology in SLE1,2. SLE is associated with a myriad of immunoregulatory abnormalities. Considerable interest has been focused on abnormalities in T cell responses or production of T cell cytokines and/or defective control by regulatory T cells. One or other of those abnormalities has been hypothesized to play an essential role in the evolution of autoimmunity and tissue pathology1. More recent data suggest, however, that an intrinsic tendency of B cells to respond excessively to immune stimulation may be an essential feature of SLE2. One feature of the B cell hypothesis is the central relationship between SLE and the production of characteristic patterns of autoantibodies, some of which are clearly involved in tissue damage. These include antiDNA in glomerulonephritis, anti-cardiolipin in thrombosis and anti-Ro in congenital heart block2. A challenge to the central role of autoantibodies in SLE, however, has come from results of genetically manipulated lupusprone MRL lpr/lpr mice that develop nephritis and vasculitis despite being unable to http://immunol.nature.com