Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Evolution of metal ions in biological systems wikipedia , lookup
Metalloprotein wikipedia , lookup
Metal carbonyl wikipedia , lookup
Spin crossover wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Coordination complex wikipedia , lookup
Stille reaction wikipedia , lookup
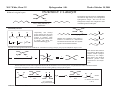
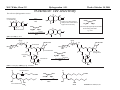
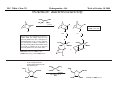
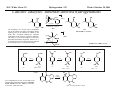
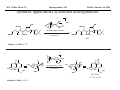
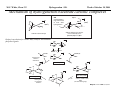
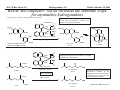
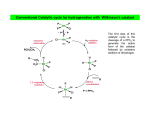
M.C. White, Chem 253 Cross-Coupling -132- Week of October 18 , 2004 Heck Reaction The Heck Reaction: L2Pd IIX2 cat. H + R'X R' + Base R Base H+ X- R Heck is stereoselective for E olefin formation Heck Org. React. 1982 (27) 345. Olefin: Increasing the db substitution dramatically decreases the rate of intermolecular Heck reactions: Base: 2o or 3o amine, NaOAc, K2CO3, KHCO3, KOAc > > X- +HNEt3 reductive elimination H LPd(II) X R R' R H L R' Pd II X L L H R PdII Pd X R'X oxidative addition L2Pd 0 β -hydride elimination (cis) H L Et2N X R' R H + PdII L R' Catalyst: Pd(II) sources often used: Pd(OAc) 2 , PdCl2PR3, PdCl2(CH3CN) Pd (0) sources: Pd(PPh 3) 4, Pd(dba)2+ PR3 NEt3 R H The base may serve a dual purpose: reducing the Pd(II) precatalyst to Pd(0) and promoting reductive elimination of the PdH(X) intermediate by shifting the equilibrium towards Pd(0). L2Pd IIX2 Neutral mechanism: coordination of olefin via dissociation of a neutral ligand. Thought to operate when X = strong σ-donor (i.e.Cl, Br or I). When aryl or vinyl halides are used, bidentate ligands can result in a partial or complete suppression of the reaction. The reaction is stereoselective for the E olefin because the corresponding TS leading to the cis olefin involves energetically unfavorable R'/R eclipsing interactions. R' = aryl, heterocyclic, vinyl, benzyl X = Br, I, OTf, Cl L reversible β-hydride elimination can lead to olefin isomerization when R= alkyl H X R' R H II Pd X II L R' internal rotation R H cis migratory also known as: olefin insertion, insertion carbopalladation Pd II X L M.C. White, Chem 253 Cross-Coupling -133- Week of October 18, 2004 Heck Reaction L2Pd II(X)2 Cationic mechanism: coordination of olefin via dissociation of a weakly associated anionic ligand. Thought to operate when X = OTf, OAc or when Ag or Tl salts (AgY or TlY; Y= CO3, OTf, OAc) are used that are capable of halide abstraction (metathesis- see Structure & Bonding -12-) NEt3 + HNO3 R Faster dissociation of the olefin leads to less β-hydride elimination. LPd R' R PdII L R' H R H H R' PdII L Pd NO3- Br II L L AgNO3 β -hydride elimination (cis) AgBr + H L internal rotation R H + NO3- L cis migratory insertion PdII L Pd II L NO3- R' R' R + H Cabri Acc. Chem. Res. 1995 (28) 2. Beletskaya Chem. Rev. 2000 (100) 3009 R' L + Pd II L X H NO3- R H H R'Br oxidative addition L2Pd 0 (II) R Et2N L Halide abstraction additives minimize db isomerization O Ph O O N I Pd(OAc)2 (10 mol%) Ph N Ph O Ph N N PPh3 (20 mol%) CH3CN, 80oC 1st product formed First example:Overman JOC 1987 (52) 4133. Grigg TL 1991 (32) 687. Pd-H insertion product I none TlOAc (1.2 eq) AgOAc (1.2 eq) 1: 2: 5 1: 0: 0 1: 0: 0 Pd-H insertion product II M.C. White, Chem 253 Cross-Coupling -134- Week of October 18, 2004 Heck: Regioselectivity of migratory insertion with neutral Pd complexes α PdII(OAc)2 1 mol% PPh3 2 mol% H Br (I) + or NEt3 or TMED (tetramethylethylene diamine) R R R β 100% 100% CO2Me 100% CN Ph 100% 99% 80% C4H9 20% CO2Me 60% 40% Ph N OCH3 100% 1% O For intermolecular Heck reactions with neutral Pd complexes and unactivated or electron-poor alkenes, the regioselectivity for R' insertion is under steric control, resulting in substitution at the less sterically hindered position. In contrast, with neutral Pd complexes and electron-rich alkenes (e.g. heteroatom substituted olefins), the regioselectivity of R' insertion is under electronic control, resulting in substitution α to the electron-donating group. Heck Org. React. 1982 (27) 345. Heck JACS 1974 (96) 1133. Hallberg Tetrahedron 1994 (50) 285. M.C. White, Chem 253 Cross-Coupling -135- Week October 18, 2004 Heck: Regioselectivity of migratory insertion with cationic Pd complexes R' α Pd II(OAc)2 P(dppp) R' H OTf + R R or Ar-X + TlOAc β 100% CO2Me R' NEt3 or iPr2NEt R 100% or 20% 60% 40% CN Ph 100% C4H9 N 100% 5% 95% OH OAc 100% O-n-Bu 80% O For intermolecular Heck reactions with cationic Pd complexes, the regioselectivity for R' insertion is predominantly under electronic control for all substrate classes. Coordination of the olefin π-system to a cationic Pd complex results in an increase in polarization of the C=C bond, and selective migration of the aryl moiety onto the carbon with lower charge density is observed. Cabri Acc. Chem. Res. 1995 28, 2-7. Cabri JOC 1992, 57, 1481-1486. Cabri Tet. Lett. 1991 32:14, 1753-1756. M.C. White, Chem 253 Cross-Coupling -136- Week of October 18, 2004 Intramolecular Heck: “exo-trig” vs “endo-trig” cyclization exo-trig endo-trig Pd L Pd X L Pd(L)n(X) X Pd(L)n(X) For the formation of small rings (5,6, or 7 membered rings) conformational effects dominate and the exo-trig mode of cyclization is generally preferred. In contrast, for the formation of macrocyclic structures (>9-membered rings), steric effects dominate and the endo-trig mode of cyclization is generally preferred. CO2CH3 CO2CH3 N Pd(OAc)2 N n Ph 3 P, Et 3 N I H 86% O N O H H N I CO2CH3 N Pd(OAc)2 O O O O O Pd(OAc)2 Tri-o-tolphosphine O Et3 N, CH 3 N N O n O CO2CH3 O I Ph 3 P, Et 3 N H 74% Overman JOC 1987 52 4130-4133. O n=3, 29% n=5, 24% n=7, 38% Stocks Tet. Lett. 1995 36:36 6555-6558. M.C. White/ M.W. KananChem 253 Cross-Coupling -137- Week of October 18, 2004 Tandem Heck: construction of adjacent quaternary C centers O I O O Bn Bn N N O I O O Pd(PPh3)2Cl2 (10 mol%) BnN Et3N, DMA, 100°C NBn O O Pd(PPh3)2 90% β -hydride elimination oxidative addition O O O The stereochemistry of the acetonide controls the Heck cyclizations such that only a single stereoisomer is observed. Despite the steric congestion of the olefins in the two cylcizations (tetra- and tri-substituted), the overall transformation proceeds efficiently. O Bn BnN I I N Pd PPh 3O BnN NBn O olefin insertion O O O I BnN PdIL O O β -hydride elimination O O Bn N BnN O Overman JACS 1999 (121) 7702. O O Bn HN I oxidative addition PdLI O olefin insertion O O I Pd BnN O L N Bn M.C. White/M.W. Kanan Chem 253 Cross-Coupling -138- Week of October 18, 2004 Intramolecular Mizoroki-Heck to construct a quaternary carbon center OMOM TfO OBOM OMOM P Pd2(dba)3 (15 mol%) OBOM Pd 0 P oxidative addition TfO - OH O OH dppb (40 mol%) Et3N, DMAc, 120°C 84% H O H OMOM reductive elimination OBOM + P Pd OMOM P OBOM Pd P In the initial insertion intermediate, there is no β-hydrogen to enable elimination of OH O H P PdII and regeneration of olefin. Et3N serves as a hydride donor, generating an H O OH H + Et2 N alkyl-hydrido species that reductively eliminates to release the desired product. OMOM OBOM OMOM OBOM + P Pd P OMOM OH OBOM OBOM O H olefin insertion + P Pd P Hirama Org. Lett. 2002 (4) 1627. O H Et2 N Pd P P Et3N OH b-hydride elimination OH H + OMOM OH Et3 N + P Pd P O H O H M.C. White/Q.Chen Chem 253 Cross-Coupling -139- Week of October 18, 2004 Tandem Heck-Hiyama Coupling OEt I EtO O O 10% Pd(OAc)2, 20% dppp R Et 3N (5 eq), H2O (2 eq), DMF, 80 °C O R Si iPr iPr R = n-Hex Pd(dppp) Isoprostanes & Neuroprostanes O 73% OH Si iPr iPr reductive elimination oxidative addition I OEt (dppp)Pd R Si iPr iPr OEt Following olefin insertion, there is no syn hydrogen available for β-hydride elimination. Instead, this intermediate is proposed to undergo a hydroxide-promoted, intramolecular Hiyama-type transmetalation followed by reductive elimination to yield the desired product. O Pd(dppp), I O R O OEt olefin insertion OEt O O intramolecular transmetalation OH I Pd (dppp) O Si iPr iPr Quan, L. G.; Cha, J.K. JACS 2002, ASAP. I R Pd OH (dppp) O Si iPr iPr R Pd OH (dppp) O I Si iPr iPr M.C. White, Chem 253 Hydrogenation -140- Week of October 18, 2004 Wilkinson’s Catalyst Wilkinson's original report: Ph3P PPh3 Rh(I) P h3P Investigations into the reactivity of (PPh3)RhCl uncovered its high activity as a homogeneous hydrogenation catalyst. This was the first homogeneous catalyst that compared in rates with heterogeneous counterparts. Cl cat. H2 (1 atm), benzene, rt quantitative Ph3P Functionality tolerated O O O OR C N OH O NO2 R Rh(I) Ph3P Compatibility with carbonyl groups indicates that the metal hydride intermediate is primarily covalent in character (lacks hydridic properties characteristic of strongly ionic M-H). See Structure & Bonding pg. 28. PPh3 H Cl cat. H H2: D2 (1:1) 50% D D Minimal H/D scrambling in the product is indicative of formation of a dihydrometal intermediate that transfers both of its hydrido ligands to the unsaturated substrate. 43.9% H D 6.1% Ethylene is not hydrogenated under these conditions but...stoichiometric hydrogen transfer from preformed dihydride complex occurs. Ph3P Rh(I) Ph3P Data indicates that formation of an ethylene/ Rh(Cl)(PPh3)3 complex inhibits hydrogen activation by the complex. This implies that dihydride formation precedes olefin complexation in the catalytic cycle. PPh 3 H Cl Ph3P cat. H Rh(III) H2 (1 atm), benzene, rt Cl rt + PPh3 Ph3P Rh(I) Ph3P PPh 3 + Cl PPh 3 The stereochemical outcome of this experiment indicates that the mechanism involves stereospecific cis hydrometallation of the unsaturated substrate followed by stereospecific reductive elimination from the resulting alkenyl (or alkyl) hydrido species. Ph3P H HO 2C H CO2H Rh (I) Ph3P PPh 3 Ph3P D D Cl D2 (1 atm), benzene 20oC Ph3P H HO 2C H CO2H meso compound major product observed Wilkinson J. Chem. Soc. (A) 1966, 1711. Rh(I) C 3H7 CH3 PPh3 Cl H 2 (50 atm), benzene 20oC H H + hexane C 3H7 CH3 cis- hexene: trans-hexene (>20:1) M.C. White, Chem 253 Hydrogenation -141- Week of October 18, 2004 Wilkinson: substrate selectivity Ph3P Rh(I) P h3P + unsaturated substrate PPh3 Cl 1 mol% H2 (1 atms.), benzene, rt unsaturated substrate + saturated substrate rate of hydrogenation of competition unsaturated substrate figure = rate of hydrogenation of 1-octene competition figure NC 14.7 HO 9.1 HO 3.4 2.6 EtO 1.8 C 3H7 , also 1-heptyne, 1-octyne C 4H 9 1.7 1.0 also, 1-decene, 1-dodecene cyclohexene Unsaturated substrates containing functionality are hydrogenated more rapidly than their unfunctionalized counterparts. The effect is suggested to result from polar functional group assisted olefin coordination to the catalyst. Terminal alkynes are hydrogenated more rapidly than terminal alkenes. This selectivity may be enhanced by use of acidic alcohol co-solvents (e.g. in benzene/ 2,2,2-trifluoroethanol, 1-hexyne: 1-octene (12:1). Terminal alkenes between C6-C12 are hydrogenated at the same rate. The same is observed for terminal alkynes. An increase in carbon chain length does not appear to affect olefin/catalyst interaction. 0.92 Conjugated dienes are reduced slower than isolated alkenes. 1,3-cyclooctadiene C 2H5 0.75 C2H5 0.71 C 2H5 C 3H7 0.69 C3H7 0.54 C3H7 Internal and branched alkenes (alkynes) are hydrogenated slower than terminal alkenes (alkynes). These differences are rationalized in terms of steric effects on olefin interaction with the catalyst and have been used to effect selective alkene hydrogenations in polyene compounds. 0.17 C 3H7 Candlin Faraday Discuss. Chem. Soc. 1968 (46) 60. M.C. White, Chem 253 Hydrogenation -142- Week of October 18, 2004 Wilkinson hydrogenation: classic dihydride mechanism PPh3 Rh P h3P (I) Ph3P oxidative addition H2 PPh3 Rh(I) Cl Ph3P Cl H H Ph3P strong π-acids (e.g. ethylene) bind tightly to the electron rich Rh center and inhibit hydrogenation Rh(I) Rh(I) Ph3P PPh3 PPh 3 Cl H2 Cl Rh(I) Ph3P S H oxidative addition PPh3 Rh(I) H H2 Rh(III) Ph3P Cl coordinatively unsaturated complex reacts w/ H2 10 4 x faster than Rh(Cl)PPh3 Ph3P H PPh3 Cl H Rh(III) Ph3P PPh3 Cl S R reductive elimination R Rh (III) Rh(III) Cl H H PPh3 Ph3P Cl +PPh 3 -PPh3 -PPh3 catalytic cycle Cl solution structure determined by NMR. PPh3 +PPh 3 Ph3P PPh3 Rh(III) H PPh3 Intermediates observed by NMR or as isolated solids in the reaction system. Formation of these "side-products" results in a reduction in the rate of hydrogenation. Ph3P PPh3 migratory insertion RDS Cl S Halpern Chem. Comm. 1973 629. Halpern J. Mol. Catal. 1976 (2) 65. Halpern Inorg. Chim. Acta. 1981 (50) 11. M.C. White, Chem 253 Hydrogenation -143- Week of October 18, 2004 Wilkinson: site selectivity Site selective hydrogenation: sterics O O Pd/C acetone, H2 (1 atm), rt 75% ketone activated cis-disubstituted O O O Ph3P O tetrasubstituted Rh(I) Ph3P highly active heterogeneous catalysts often cannot achieve high levels of selectivity. H Rh(I) Ph3P PPh3 PPh3 Cl Chlorotris(triphenylphosphine)rhodium I Cl 1 mol% H2 (1 atm), benzene/EtOH, rt 95% Strem catalog 2001-2003 1g = $42 O Ph3P O O Pedro JOC 1996 (61) 3815. Site selective hydrogenation: sterics and electronics H3 CO H3 CO H O O H O trisubstituted O HO CH(CH3 )C2 H5 O HO O O MeO CH(CH3 )C2H5 H O H O O MeO cis-disubstituted Ph3P O conjugated diene OH O Rh (I) PPh3 Ph3P Cl ~30 mol% H 2 (1 atm), tol, rt 92% H trisubstituted O O O OH H only site of hydrogenation O H Fisher J. Med. Chem. 1980 (23) 1134. Ivermectin H OMe OMe cis vs. trans-disubstituted olefins: O cis-disubstituted O CO2Me Ph3P Rh(I) CO2Me PPh3 Ph3P HO trans-disubstituted OAc PGE 2 Cl cat. H2 (1 atm), benzene/acetone, rt 80% HO OAc PGE 1 Schneider JOC 1973 (38) 951. M.C. White, Chem 253 Hydrogenation -144- Week of October 18, 2004 Wilkinson: diastereoselectivity Ph3P Rh(I) PPh3 Ph3P Cl 1 mol% H2 (1 atm.), benzene/EtOH, rt H Me R = H : 73% endo R = Me : 92% endo Me R R R endo Rationale for observed diastereoselectivity: Olefin binds the catalyst from the least sterically hindered exo face. Subsequent cis hydrometallation of the exo face followed by stereospecific reductive elimination of the alkyl metal hydrido intermediate results in overall cis addition of H2 to the least sterically hindered exo face of the olefin. exo H H H Ph3P Ph3P Rh(III) PPh3 Cl H Rh(III) PPh3 ClR vs. Rousseau J. Mol. Cat. 1979 (5) 163. Jardine Prog. Inorg. Chem. 1981 (28) 63. R olefin complexation and hydrogenation from sterically less hindered face BnO OMOM BnO OBn Ph3P Rh(I) PPh 3 BnO P h3P Cl 30 mol% H2 (1 atm.), tol, rt 83% OMOM BnO OBn Lowary OL 2000 (2) 167. M.C. White, Chem 253 Hydrogenation -145- Week of October 18, 2004 Wilkinson: directing group effects Ph3P OH Rh(I) PPh3 Ph3P Cl 0.04 mol% H 2 (6.8 atm, 100psi), benzene, 50oC OH no reaction Ph3P trisubstituted MeO K+ B - Ph3P Rh(I) note: when Pd/C was used a mixture of cis and trans isomers resulted PPh3 H Cl 0.04 mol% MeO H 2 (6.8 atm, 100psi), benzene, 50oC cis isomer (exclusive) 68% PPh3 O -K+ O H Rh PPh3 H H trisubstituted MeO The slow reaction without the alkoxide is attibuted to the steric hinderance of the tri-substituted double bond, which renders it less able to coordinate to the Rh. The protonated alcohol is not a strong enough nucleophile to associatively displace the anionic chloride ligand. Base-assisted formation of the alkoxide results in effective displacement of the chloride ligand and thus directs olefin complexation from the same face. Thompson JACS 1974 (96) 6232. Jardine Prog. Inorg. Chem. 1981 (28) 63. M.C. White, Chem 253 Hydrogenation -146- Week of October 18, 2004 Schrock- Osborn /Crabtree: Cationic catalysts Diene ligated cationic catalysts mode of activation: PCy3 Ir(I) + + H (PF 6-) Ir(III) H2 N PCy3 (PF 6- ) H cis-oxidative addition cis-migratory insertion N diene ligated catalyst precursor + Ir(III) PCy3 (PF 6- ) H N + S Ir(I) S PCy3 + (PF 6- ) repeat Ir(I) N S PCy3 (PF 6- ) cis-reductive elimination N solvated active catalyst Crabtree Acc Chem Res 1979 (12) 331. Wilkinson's catalyst P h3P Rh (I) Turnover Frequency (TOF) PPh3 Ph3P Cl benzene/EtOH, 25oC 650 700 13 ---- 4000 10 ---- ---- 6400 4500 3800 4000 + Schrock-Osborn catalyst Rh (I) PPh3 (PF 6- ) PPh3 CH2Cl2, 25oC + Crabtree's catalyst Ir(I) PCy3 N (PF 6- ) CH2Cl2, 25oC TOF = mol reduced substrate/mol catalyst/h "Coordinatively" unsaturated cationic hydrogenation catalysts are the most active homogeneous hydrogenation catalysts developed thus far. Use of weakly coordinating solvents provides the olefin substrate with relatively free access to the metal's reactive site. These cationic catalysts are also remarkably selective.... M.C. White, Chem 253 Hydrogenation -147- Week of October 18, 2004 Cationic catalysts: substrate-directed hydrogenations + OH PCy3 Ir(I) OH OH (PF6 -) N 2.5 mol% H CH2Cl2, H 2 (1 atm), rt Me H Me Me 98% The availability of a second "open" coordination site on the catalyst now makes it possible to bind a ligating group on the substrate in addition to the olefin. This "two-point" binding has important implications on the selectivity of product formation. The ability of a late metal complex to effectively bind hard functionality (hydroxyls, ketones, etc...) is attributed to the lewis acidic properties imparted on the complex by the overall positive charge. 64:1 Py Cy3P Pd/C (EtOH), 1:5 (sterics) + H (PF6-) Ir(III) H OH Me Crabtree JOC 1986 (51) 2655. i-Pr Other functionalities with lewis basic sites also direct: Esters: Ketones: Ethers O CO2Me Me Me Me above >99% Me Me H 56:1 Pd/C 1.35:1 Amides: Me (±) 124:1 Pd/C 1.26:1 O N H H Me H above 5 mol% O O above >99% Me N For a comprehensive review of cyclic and acyclic substrate-directed hydrogenations see: Hoveyda, Evans, and Fu Chem. Rev. 1993 (93) 1307 and D.A. Evans; Chem 206 notes. Me O above 97% (±) O CO2Me Me Me (±) 999:1 Pd/C 1:4 O N N H O >99:1 Pd/C 1:9 (steric approach control) H M.C. White/Q. Chen Chem 253 Hydrogenation -148- Week of October 18, 2004 High catalyst loadings: diminished yields and selectivities + OH Ir( I) PCy3 (PF6 -) OH OH N Me CH2Cl2, H2 (1 atm), rt H H Me Me A A decrease in selectivity is observed at higher catalyst loadings. It is possible that higher catalyst loadings promote the formation of dimeric (Crabtree suggested M-H-M) species that no longer have the "open" coordination site necessary for providing effective directing effects in olefin hydrogenation. No experimental data exists thus far to support this hypothesis. Dimished yields are observed with higher catalyst loadings. This can be rationalized on the basis that higher catalyst loadings promote the irreversible trimerization of the coordinatively unsaturated catalysts to yield inactive triiridium hydride bridged complexes. Such complexes have been isolated by Crabtree from reaction mixtures of more sterically hindered olefins that did not proceed to completion. Crabtree Acc. Chem. Res. 1979 (12) 331. B yield selectivity (ratio A:B) 2.5 mol% 99% 139:1 20 mol% 48% 74:1 Stork JACS 1983 (105) 1072. Crabtree JOC 1986 (51) 2655. M.C. White, Chem 253 Hydrogenation -149- Week of October 18, 2004 Synthetic applications of directed hydrogenations + Ph2 P OH ()n= 3 P Ph2 O O Me OH Rh (I) MOMO MOMO (BF4-) O [Rh(NBD)(DIPHOS-4)]+BF4- H OSEM MOMO O NaH, THF H2 (800 psi), rt O MOMO Me H OSEM H O 68% Paquette OL 2002 (4) 937. + Ir(I) H H Me O HO OH (PF 6-) HO HO Me N [Ir(COD)(py)(PCy3)]+PF6- O Me PCy3 H2, CH2Cl2 H H Me 10 Me O H HO Me O HO OH HO 99% Yield, d.r. 11:1 at C10 Barriault OL 2001 (3) 1925. M.C. White, Chem 253 Hydrogenation -150- Week of October 18, 2004 Mechanism of hydrogenation:bidentate cationic complexes + PPh 3 Rh(I) (PF 6- ) nbd (norbornadiene) ring strain results in more facile hydrogenation diphos + Ph 2 P (PF 6-) Rh(I) () n= 3 P Ph 2 PPh3 Schrock-Osborn type catalyst most commonly used: [Rh(nbd)(diphos-4)]BF4 Schrock-Osborn catalyst Halpern's mechanism for cationic Rh(I) catalysts with bidentate phosphine ligands: + Ph2 P Rh(I) P Ph2 H2 MeOH Ph CO 2Me NHAc stereospecific reductive elimination H + Ph2 P Rh(I) P Ph2 + Ph Ph CO 2Me (R) S NHAc S observed by NMR H Ph 2 P Rh P Ph2 Ph2 P H R NH (III) P Ph2 + O H Ph H S observed by NMR stereospecific cis-migratory insertion Ph 2 P P Ph 2 Rh(I) R O NH observed by NMR Rh (III) R H O + H Ph H2 NH oxidative addition (OA) RDS (rate determining step) Halpern Science 1982 (217) 401. M.C. White, Chem 253 Hydrogenation -151- Week of October 18, 2004 Mechanism of monodentate cationic complexes Halpern notes that the hydrogenation mechanism for bidentate ligated cationic complexes where olefin substrate coordination precedes oxidative addition of H 2 may not be operating for cationic catalysts with monodentate ligands. Schrock-Osborn invoke involvement of the dihydride complex (below) in the principle hydrogenation pathway for their catalyst. Halpern notes some significant differences in the reactivities towards H2 of the catalysts w/ bidentate and monodentate phosphine ligands. + In the absence of olefin substrate, no further uptake of H 2 can be detected. The only species observed by NMR is the cationic, 4-coordinate solvated species. Ph 2 P Rh(I) S H2 P Ph 2 Rh (I) S + H + Ph2 P k1 S Rh(I) P Ph 2 H P Ph 2 k-1 Ph 2 P S only species observed by NMR PPh3 Rh(I) S H2 PPh3 Rh (I) PPh3 PPh 3 Rh (III) H S k1 P h3P S Ph3P + H + + Treatment of the monodentate catalyst with H 2 resulted in detection of the Rh(III)-dihydride complex. k-1 S only species observed by NMR Halpern JACS 1977 (99) 8055. Schrock & Osborn JACS 1976 (98) 2135. The Trans Effect: To explain the difference in reactivities towards H2 of the catalysts, Halpern invokes the trans effect. The trans effect is defined as the labilization of ligands trans to certain other ligands. The trans effect often arises when a ligand shares an orbital with another ligand of strong σ-bonding character. Because phosphine forms a strong σ bond with Rh, trans Rh-H bonds formed will be weak because the orbital is not as available for bonding to H. In the case of the bidentate complex, cis addition of H2 requires that one hydride share an orbital with a phosphine. Since both hydride and phosphine are strong σ-bonding ligands, the dihydride adduct, once formed, is highly unstable and thus rapidly reverts back via reductive elimination to the solvated 4-coordinate species. In the case of the monodentate phosphine complex, a H2 adduct can form where neither H ligand is trans to a phosphine. Classic example of the trans effect: synthesis of "cis-platinum" a chemotherapeutic agent Cl Cl Pt(II) Cl Cl 2NH3 Cl Pt(II) Cl - NH3 Cl Cl has a stronger "trans influence" than NH3 NH3 Cl Cl (II) NH3 Pt NH3 only the cis isomer is formed H 3N H 3N Pt(II) NH3 NH3 2+ Cl H 3N H 3N Pt(II) Cl + Cl NH3 Cl has a stronger "trans influence" than NH3 H 3N Pt(II) Cl NH3 Cl only the trans isomer is formed M.C. White, Chem 253 Hydrogenation -152- Week of October 18, 2004 Asymmetric Hydrogenation A bidentate, C2 symmetric version of the cationic Schrock-Osborn catalyst affords extraordinarily high levels of enantioselectivity in the hydrogenation of achiral enamides. This was the first demonstration that a chiral transition metal complex could effectively transfer chirality to a non-chiral substrate with selectivities that rival those observed in enzymes. Recall that this led to the 1st commericalized asymmetric process using a chiral transition metal complex: Monsanto Process for the industrial production of L-DOPA (see Structure and Bonding, pg. 4) + + Rh(I) PPh3 MeO (PF 6- ) Rh(I) P (PF 6-) P PPh 3 OMe DIPAMP (common name for this bidentate chiral phosphine ligand) Schrock-Osborn catalyst CO2H CO2H NHAc Knowles JACS 1975 (97) 2567. i-PrOH, H 2 (1 atm), rt >99% yield NHAc 93% ee A variety of bidentate chiral phosphines have since been synthesized and used to effect the hydrogenation of aromatic enamides (important substrates for the efficient generation of amino acids): H PPh2 PPh2 PPh2 O PPh2 NMe2 Fe PPh2 PPh2 O PPh2 PPh2 PPh2 PPh2 H SKEWPHOS (92% ee) Chiraphos (99% ee) NORPHOS (95% ee) BPPFA (93% ee) DIOP (85% ee) R PPh2 P R R PPh2 P BINAP (100% ee) R DuPHOS (99% ee) H H PPh 2 PPh 2 BICP (97% ee) We'll see these ligands again effecting asymmetry in a wide assortment of mechanistically unrelated metal catalyzed reactions with prochiral substrates. PCy2 "Privileged ligand class": ligands that communicate asymmetry effectively with a transition state Fe PPh2 localized at the metal center, irrespective of the nature of the transition state. PPh2 E.N. Jacobsen; personal communication JOSIPHOS (96% ee) E. N. Jacobsen. Chem 153 notes. Spring 2001. For review on DuPhos: Burk Acc. Chem. Res. 2000 (33) 363. M.C. White, Chem 253 Hydrogenation -153- Week of October 18, 2004 Origin of Asymmetric Induction + (ClO4-) H 3C It was concluded from kinetic measurements that the minor diastereomer was 580 fold more reactive towards H2 oxidative addition (recall the RDS at rt). This factor offsets its lower concentration in solution and results in a 60:1 product ratio in favor of the R enantiomer. P Rh(I) P H 3C (S,S-CHIRAPHOS) (R) CO2Et CO2H H2 (1 atm), 25oC, MeOH NHAc NHAc N-acetyl-(R)-phenylalanine >95% ee stereospecific H migratory insertion / stereospecific H/C reductive elimination both to the olefin face bound to Rh HN CO 2Me Rh(I) Ph O + + (ClO4-) (ClO4-) CH3 NH P P P MeO2 C H 3C Rh(I) CH3 H 3C P O Ph olefin bound to Rh via its si-face olefin bound to Rh via its re-face major diastereomer formed in solution (identified by NMR and x-ray crystallography) minor diastereomer formed none detected by NMR (must be less than 5% present in solution) Halpern Science 1982 (217) 401. M.C. White/Q. Chen Chem 253 Hydrogenation -154- Week of October 18, 2004 Crystal structure of major diastereomer + (ClO4-) HN CO 2Me Rh(I) Ph O CH3 P P CH3 olefin bound to Rh via its re-face major diastereomer formed in solution (identified by NMR and x-ray crystallography) Major enantiomer observed upon exposing crystal to H2: (R) CO2Et NHAc Minor enantiomer observed upon exposing crystal to H2. (S) CO2Et NHAc N-acetyl-(R)-phenylalanine >95% ee Halpern Science 1982 (217) 401. M.C. White/Q. Chen Chem 253 Hydrogenation -155- Week of October 18, 2004 Monohydride catalysts: RuClH(PPh3)3 Wilkinson's original report: "In contrast to the rhodium system, ethanol plays an intimate part in the hydrogenation mechanism; in the absence of such a co-solvent, hydrogenation is exceedingly slow." Wilkinson Nature 1965 (208) 1203. RuCl2(PPh3) 2 cat. H2 (1 atm), benzene:ethanol, rt quantitative The active species was identified as the monohydride, thought to form via heterolytic cleavage of H2, with ethanol acting as a base. The monohydride can also be prepared in 100% benzene if an equivalent of NEt3 is added. One mole of H 2 is absorbed with respect to Ru and amine hydrochloride is quantitatively formed. Wilkinson J. Chem. Soc. (A) 1968 3143. H RuCl2(PPh3) 3 H2 (1 eq) NEt3 (1 eq), benzene Ph3P PPh3 Ru(II) + - Cl +HNEt3 RuCl(H)(PPh3)3, highly distorted trigonal bipyramidal. Skapski Chem. Comm. 1968 1230. PPh3 Cl Effect of base on conversion O O RuCl2(PPh3) 2 cat. H2 (126 atm), base (1 eq) benzene, 40oC, 6h O O Base Tsuneda Bull. Chem. Soc. Jpn. 1973 (46) 279. NEt3 Et2NH BuNH2 aniline Ca2CO3 Na 2CO3 none % Conversion 95.4 95.4 86.5 88.1 95.2 73.0 76.0 + fully satuturated products M.C. White, Chem 253 Hydrogenation -156- Base promoted heterolytic cleavage: Week of October 18, 2004 Mechanism of H2 Activation H σ-donation>> π-backbonding Mn Mn heterolytic cleavage H σ-complex generally observed for electrophilic metals that are in their highest stable oxidation state within the context of their ligand framework. H + BH+ note: there is no oxidation state change at the metal Example: + (BF 4) N Ir(III) + (BF4) - NH2 L H δ+ N L Ir(III) H L NH3+ H L H H 1 2 Complexation of dihydrogen to the electrophilic, cationic Ir(III) center is predominantly σ-donating in nature. Donation of electron density from the H-H σ-bond to an empty Ir orbital leaves the H-H with a partial positive charge. The pendent NH2 group is thought to act as an internal base effecting heterolytic cleavage of the acidified dihydrogen σ-complex via deprotonation.When L = PPh3, the equilibrium lies far to the right and only the dihydride 2 is observed. When more basic alkyl phosphines are used (L= PBu3) the equilibrium lies to the left with the H2 complex 1 being observed exclusively by NMR. It was hypothesized that moving to a more basic phosphine increases the electron density at the metal center. This makes the metal a less effective σ-acceptor and attenuates its ability to effectively acidify the dihydrogen complex. Crabtree Chem. Commun. 1999 297. σ-bond metathesis: the base is effectively one of the ligands on the metal Cl RuCl2(PPh 3)3 H2 δ+ H external amine base may still drive the rxn forward by forming insoluble amine hydrochloride salts δ− HCl RuCl(PPh 3)3 H σ-bond metathesis Crabtree The Organometallic Chemistry of the Transition Metals: 3rd Edition; Wiley: New York; 2001. RuHCl(PPh3)3 M.C. White, Chem 253 Hydrogenation -157- Week of October 18, 2004 BINAP-Ru complexes: Noyori increases the substrate scope for asymmetric hydrogenations The first report: asymmetric hydrogenation of (Z)-enamides Interestingly, E-enamides are completely unreactive towards these hydrogenation conditions. No rationale for this has been presented. air-sensitive O Ph 2 P H3CO OAc H3CO HO O P Ph 2 NCOCH3 AcO O Ru(II) NCOCH3 AcO O O OAc (R)-1 (0.5-1 mol%) HO EtOH:CH2Cl2 (5:1), H2 (4 atm), 23oC Noyori JACS 1986 (108) 7117. Noyori ACIEE 2002 (41) 2008. NCH3 OCH3 OCH3 H morphine 92% yield 95% ee Asymmetric hydrogenation of allylic and homoallylic alcohols: Tol2 P P Tol2 E-olefin OH O O Ru(II) regioselectivity: allylic and homoallylic alcohols are hydrogenated whereas bis homoallylic and higher analogues are left untouched. O O (S)-1 0.01 mol% MeOH, H2, 18-20oC geraniol 97 to >99% yields OH (R)-citronellol allylic olefin geometry may dictate the stereochemical outcome of the hydrogenation. Practical consequence: to obtain high ee's must start with stereochemically pure olefins. 96% ee Z-olefin (S)-1 0.2 mol% MeOH, H2, 18-20oC nerol OH OH (S)-citronellol 98% ee Noyori JACS 1987 (109) 1596. M.C. White, Chem 253 Hydrogenation -158- Week of October 18, 2004 BINAP-Ru complexes: Noyori increases the substrate scope for asymmetric hydrogenations The first demonstration of high asymmetric induction in the hydrogenation of substrates lacking an acylamino group: asymmetric hydrogenation of α,β-unsaturated carboxylic acids CO2H Ph 2 P H 3C (S) O Ru CH3 (II) H3C O P Ph 2 CO2H CO2H O O CH3 H2 (4 atm): 91% ee H2 (101 atm): 50% ee (S)-1 (0.5-1 mol%) MeOH, H2, CO2H 15-30oC The degree of asymmetric induction is significantly affected by the H 2 pressure in a substrate specific manner. The implication of this is that a range of H2 pressures must be screened to achieve optimal asymmetric induction on a substrate by substrate basis. No trend was observed and no rationale for the emperical observation was given. (S) Ph H2 (4 atm): 48% ee H2 (112 atm): 92% ee Asymmetric Synthesis of (S)-Naproxen: CO2H (S)-1 (0.5 mol%) MeOH, H2 (135 atm), H3CO 92% yield 97% ee CO2H 15-30oC H 3CO (S)-Naproxen Noyori JOC 1987 (52) 3174. M.C. White, Chem 253 Hydrogenation -159- Week of October 18, 2004 Mechanism of BINAP-Ru hydrogenation of α,β-unsaturated acids Ph 2 P P Ph 2 O Ru (II) O O O 1 CO 2H note: mechanism is valid for both enantiomers of BINAP. No rationalization for the enantiofacial selectivity is given. AcOH Ph 2 P CO 2H P Ph 2 CO 2H O O Ru (II) O H2 O Heterolytic cleavage of H2 RDS H+ Ph 2 P P Ph 2 O Ru (II) Ph 2 P O O P Ph 2 O Ph 2 P protonolysis P Ph 2 H+ Ru (II) O O O H note: no oxidation state change to the metal O Ru (II) O O cis-migratory insertion O H O Reactions in MeOD CO2H H 3C CH3 D (S)-1 H2, MeOD H 3C H H CH3 CO2H Experiment indicates that the hydrogen α to the acid comes from H2 whereas the β-hydrogen comes from MeOH. Regio- and stereospecific deuterium incorporation indicates that cis-migratory insertion of the Ru-H is stereospecific as is cleavage of the Ru-C bond via protonolysis. The lack of D incorporation into the α position indicates that the rate of H/D exchange between the Ru-H and solvent is slow. Halpern JACS 1991 (113) 589. M.C. White, Chem 253 Hydrogenation -160- Week of October 18, 2004 Question of the Week Ru(CH3CO2) 2-[(S)-BINAP] catalyzes the hydrogenation of α-(acylamino)acrylic esters to give the (S) saturated product in >90% ee's. Propose a mechanism that accounts for the observed mixture of hydrogenation products when the reaction is run in MeOD. Note: your mechanism need not rationalize the absolute stereochemistry obtained. CH3 O Ph NH Ph 2 P P Ph 2 O Ru (II) O O Ph O O O O CH3 CH3 CH3 NH Ph NH Ph NH (S)-1 H O MeO H2 (1 atm), MeOD H H O H OMe H H O D OMe 79:14:2 H D O H OMe