Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Rheinische Friedrich-Wilhelms-Universität Bonn
Landwirtschaftliche Fakultät
&
Technische Universität München
Fachgebiet für Wildbiologie und Wildtiermanagement
Diplomarbeit
Effects of habitat fragmentation and isolation on the
genetic variability of khulan (Equus hemionus) in
Mongolia
vorgelegt von
Stephanie Pietsch
Februar 2007
Prüfer:
PD Dr. Ralph Kühn
Fachgebiet für Wildbiologie und Wildtiermanagement
Technische Universität München
Prof. Dr. K. Schellander
Institut für Tierzucht und Tierhaltung
Universität Bonn
Betreuer:
Dipl.-Biol. Bernhard Gum & PD Dr. Ralph Kühn
Fachgebiet für Wildbiologie und Wildtiermanagement
Abstract
Abstract
Habitat fragmentation and isolation caused by anthropogenic activities are the major factors
for wildlife population and species extinction worldwide. Thus, assessing the species-specific
effects of habitat fragmentation on population genetic structure is important to evaluate the
viability of wildlife populations in fragmented landscapes. In this study, the Mongolian
khulan (E. hemionus) was exemplarily used to investigate the effects of human disturbance on
the genetic variability as well as the level of gene flow and population differentiation. Eighty
samples were collected in the three main distribution areas (Great Gobi A, Gobi B and Small
Gobi) of Equus hemionus in southern Mongolia. DNA was extracted from different biological
material (bones, tissue and faeces) and cross-amplified at eleven equid microsatellite loci.
Reliable noninvasive genetic monitoring of khulan populations was enabled using (i) a PCRRFLP analysis for species identification and (ii) a quantitative real-time PCR assay for the
evaluation of DNA quality and quantity. The levels of intra- and interpopulation genetic
diversity were investigated with different population genetic methods like F-statistics,
AMOVA, demographic analysis and Bayesian population assignment. In order to detect
potential spatial population boundaries, we used the ´isolation by distance` model and the
Monmonier`s maximum difference algorithm. The statistical analysis revealed a detectable
population differentiation of khulans in the study area presumably due to human disturbance
in terms of poaching and increased livestock competition. So far the population fragmentation
does not affect the genetic diversity within the khulan populations. These findings support the
hypothesis that anthropogenic barriers act as a moderator of gene flow because of a high
resistance to khulan movements, and hence their cumulative effect has led to the
differentiation of two genetic units.
I
List of Abbreviations
List of Abbreviations
A
average number of alleles per locus
AMOVA
hierarchical analysis of molecular variance
AP
private alleles
AR
mean allelic richness per population
BLAST
Basic Local Alignment Search Tool
bp
base pair
BSA
bovine serum albumin
°C
degrees Celsius
Ct
cycle threshold
cyt b
cytochrome b
DNA
deoxyribonucleic acid
dNTPs
deoxynucleotide triphosphate
EtOH
ethanol
Exon
expressed region
Fig.
figure
FST
fixation index
GA
Gobi A
GAPDH
glyceraldehyde-3-phosphate dehydrogenase
GB
Gobi B
GPS
Global Positioning System
GS
Small Gobi
h
hour
He
expected heterozygosity
Ho
observed heterozygosity
HWE
Hardy-Weinberg equilibrium
IAM
infinite allele model
IBD
isolation by distance model
k
number of alleles
K
number of subpopulations
km
kilometre
LD
linkage disequilibrium
LINE
long interspersed elements
log
logarithm
II
List of Abbreviations
M
mole
MCA
melting curve analysis
MCMC
Markov chain Monte Carlo simulation
mg
milligram
MgCl2
Magnesium chloride
min
minute
ml
millilitre
mM
millimole
mRNA
messenger ribonucleic acid
mtDNA
mitochondrial deoxyribonucleic acid
N
number
NaOH
sodium hydroxide
Ne
effective population size
NGS
noninvasive genetic sampling
NTC
non template control
PCR
polymerase chain reaction
pg
picogram
PHW
Hardy-Weinberg probability test
q
average proportion of membership
qPCR
quantitative PCR
r
range of allele size
RFLP
restriction fragment length polymorphism
s
second
SINE
short interspersed elements
SMM
stepwise mutation model
SPA
strictly protected area
TPM
two-phase model
u
units
V
volt
µ
mutation rate
µl
microlitre
µm
micrometre
µM
micromole
III
Table of Contents
Table of Contents
Abstract................................................................................................. I
List of Abbreviations ..........................................................................II
List of Figures ................................................................................... VI
List of Tables ....................................................................................VII
1 Introduction....................................................................................1
2 Materials and Methods .................................................................4
2.1
2.2
2.3
2.4
Study site ................................................................................................. 4
Study species ........................................................................................... 5
Sampling and sample preservation ........................................................ 5
Laboratory procedures............................................................................ 5
2.4.1
2.4.2
2.4.3
2.4.4
2.4.5
2.4.6
2.5
Statistical analysis................................................................................. 14
2.5.1
2.5.2
2.5.3
2.5.4
2.5.5
3
Intrapopulation genetic diversity.................................................................. 14
Interpopulation genetic diversity .................................................................. 14
Demographic analysis .................................................................................... 14
Bayesian population assignment ................................................................... 15
Spatial analysis ............................................................................................... 15
Results ...........................................................................................17
3.1
3.2
3.3
3.4
Species identification ............................................................................ 17
Quantitative PCR .................................................................................. 18
Microsatellite amplification and reliable genotyping ......................... 21
Statistical analysis................................................................................. 23
3.4.1
3.4.2
3.4.3
3.4.4
3.4.5
4
DNA extraction ................................................................................................. 5
Species identification........................................................................................ 7
Quantitative PCR ............................................................................................. 9
Microsatellite amplification........................................................................... 10
Contamination control ................................................................................... 13
Genotyping criteria and analysis .................................................................. 13
Intrapopulation genetic diversity.................................................................. 23
Interpopulation genetic diversity .................................................................. 24
Demographic analysis .................................................................................... 25
Bayesian population assignment ................................................................... 26
Spatial analysis ............................................................................................... 29
Discussion .....................................................................................30
4.1
4.2
4.3
Species identification ............................................................................ 30
Quantitative PCR and reliable genotyping.......................................... 32
Statistical analysis................................................................................. 33
4.3.1
4.3.2
4.3.3
4.3.4
4.3.5
Intrapopulation genetic diversity.................................................................. 33
Demographic analysis .................................................................................... 34
Interpopulation genetic diversity .................................................................. 34
Bayesian population assignment ................................................................... 35
Spatial analysis ............................................................................................... 35
5 References.....................................................................................37
APPENDIX I ......................................................................................46
IV
Table of Contents
APPENDIX II.....................................................................................47
APPENDIX III ...................................................................................48
APPENDIX IV ...................................................................................52
APPENDIX V.....................................................................................53
APPENDIX VI ...................................................................................56
APPENDIX VII..................................................................................61
6 Acknowledgments........................................................................66
V
List of Figures
List of Figures
Fig. 1: Map of the three study areas ....................................................................................... 4
Fig. 2: Sequence alignment of a 335bp fragment of the mitochondrial cyt b gene ............ 9
Fig. 3: RFLP banding patterns of an amplified 335 bp fragment of the cyt b gene
obtained from two different Mongolian equid species................................................ 18
Fig. 4: Plot of the known initial DNA concentration........................................................... 18
Fig. 5: Melting curve analysis of the standard dilution series from equid GAPDH gene
amplified via qPCR. ....................................................................................................... 19
Fig. 6: Distribution of DNA concentration .......................................................................... 20
Fig. 7: Categorization of DNA concentrations calculated via qPCR................................. 20
Fig. 8: Genotyping electropherogram patterns of the 6-fold DNA dilutions series ......... 22
Fig. 9: AMOVA analysis conducted for three populations in one group.......................... 24
Fig. 10: Bayesian clustering results of STRUCTURE ........................................................ 26
Fig. 11: Results of the assignment test (GENECLASS)...................................................... 27
Fig. 12: The means (averaged over posterior probabilities) of migration rates between
khulan subpopulations as detected by BAYESASS. ................................................... 28
Fig. 13: Mantel test on isolation by distance between khulan populations....................... 29
Fig. 14: Detection of genetic boundaries .............................................................................. 29
VI
List of Tables
List of Tables
Table 1: Characterization of the analysed equine microsatellite loci in Mongolian khulan
(E. hemionus). ................................................................................................................. 12
Table 2: DNA concentrations were calculated from the slope and Y-intercept of the
trend line from the standard curve............................................................................... 19
Table 3: Microsatellite diversity indices of khulan (Equus hemionus) populations. ....... 23
Table 4: Matrix of pairwise of FST values (Weir and Cockerham 1984) between the
khulan populations in Mongolia. .................................................................................. 24
Table 5: AMOVA analysis performed for three group-combinations .............................. 25
Table 6: Bayesian clustering results of STRUCTURE for the assignment of individuals
to each subpopulation .................................................................................................... 27
VII
Introduction
1 Introduction
Habitat fragmentation and isolation caused by anthropogenic activities are the major factors
for population, metapopulation and species extinction worldwide (Wilcox and Murphy 1985).
Habitat loss in particular can limit gene flow between populations (Hedrick 1995). This
reduced connectivity is suspected to increase inbreeding (Saccheri et al. 1998) and accelerate
the loss of genetic diversity because of genetic drift (Frankel and Soulé 1981). In addition
these processes limit the evolutionary potential to adapt to environmental changes (Lande
1998, Fraser and Bernatchez 2001). Consequently, in conservation biology and wildlife
management, the species-specific effects of habitat destruction on population genetic structure
and viability are the most important contemporary conservation issue (Lacy 1987, Wiens
1996).
The Asiatic wild ass (khulan: Equus hemionus), once distributed across Central Asia,
continuously declined in both numbers and range during the 19th century (Reading et al.
2001). In the IUCN Equid Action Plan the status of Equus hemionus is classified as
“insufficiently known” and the species is listed as vulnerable (IUCN 1996; Feh et al. 2002).
Currently, the Gobi Desert region in southern Mongolia holds an estimated 20,000 khulan
with distribution restricted to the five Strictly Protected Areas (SPAs) Great Gobi A, Great
Gobi B, Gobi Gurvan Saikhan, Small Gobi A and Small Gobi B (Mongolian Ministry of
Nature and Environment 2003). The largest free-ranging khulan populations have received
full protection by law in Mongolia since 1953 (Reading et al. 2001). Deterministic factors for
recent regional declines include poaching for meat (Duncan 1992), increased inter-specific
competition with livestock for resources (Zhirnov and Ilyinsky 1986) and habitat barriers like
fenced borders and railroads (Kaczensky and Walzer 2003).
Unfortunately, logistic and technical difficulties of comprehensive traditional field monitoring
(e.g. via radiotelemetry) impede the investigation of wildlife population dynamic and genetic
consequences of habitat fragmentation across large spatial and temporal scales (Tishendorf
and Fahrig 2000, Kaczensky and Walzer 2003).
However, using highly variable genetic markers like microsatellites (Goldstein and
Schlötterer 1999) along with noninvasive sampling methods (Taberlet and Bouvet 1992,
Morin and Woodruff 1996), researchers can monitor free-ranging endangered wildlife
populations without capture or direct handling of individual animals.
1
Introduction
Various materials, either (i) left behind from the living animal, (ii) derived from skeletal
remains of carcasses or (iii) obtained from museum specimens provide valuable sources of
DNA, e.g. faeces (Flagstad et al. 1999), urine (Valiere and Taberlet 2000, Hausknecht et al.
in prep.), hair (Taberlet and Bouvet 1992), sloughed skin (Valsecchi et al. 1998), bones
(Hardy et al. 1994), teeth (Wandeler et al. 2003) and horn (Worley et al. 2004).
Several studies illustrated the great potential of noninvasive genetic sampling (NGS) and its
application to a wide array of wildlife research topics such as the detection of rare species
(Valiere et al. 2003), population size and habitat use (Kohn et al. 1999), individual
identification and sexing (Murphy et al. 2003), genetic diversity and gene flow (Fernando et
al. 2000) and social structure (Garnier et al. 2001).
Despite its great potential, recent controversies have demonstrated potential pitfalls and
limitations of NGS. Genotyping errors such as allelic dropout (Navidi et al. 1992, Taberlet et
al. 1996, 1999) and false alleles (Goossens et al. 1998; Bradley and Vigilant 2002) are
reported to reduce the reliability and accuracy of microsatellite genotyping data. Genotyping
errors can lead to misinterpretation of genotypes (Taberlet et al. 1999) and thus bias the
inferred biological results (Creel et al. 1999).
Consequently, wildlife scientists suggest strategies for reliable microsatellite genotyping
based on systematic tracking of the causes and consequences of genotyping errors (Taberlet
and Luikart 1999, Bonin et al. 2004, Broquet and Petit 2004, Pompanon et al. 2005). Stringent
guidelines have been established, either to reduce genotyping errors, e.g. the multiple tube
approach (Taberlet and Luikart 1999) and prescreening of template DNA via qPCR (Morin et
al. 2001), or to avoid misinterpretation of genotypes, e.g. the pairwise mismatching method
(Paetkau 2003), the maximum likelihood approach (Miller et al. 2002) and statistical
computer simulations (Taberlet et al. 1996, Valiere et al. 2002).
The implementation of a reliable and feasible NGS method contributes to verify the working
hypothesis of this study:
Habitat fragmentation and potential population isolation have already led to a
detectable genetic differentiation among khulan populations in southern Mongolia.
This hypothesis is based on the results of a radio-telemetry study on free-ranging khulans in
southwest Mongolia conducted by Kaczensky et al. (2003). They observed reduced movement
patterns of khulans between the SPAs Gobi A and B. Khulan populations in the Gobi B are
supposed to be isolated.
In order to particularly interpret population genetic structure across different geographical
scales, a new scientific field, landscape genetics, provides a powerful tool (Manel et al. 2003,
2
Introduction
Scribner et al. 2005). The basic principle of landscape genetics is to delineate spatial
population boundaries like clines (Sokal 1998), metapopulations to gene flow (Hanski 1998),
isolation by distance (Cassens et al. 2000), genetic barriers to gene flow (Piertney et al. 1998)
and random patterns (Piglucci and Barbujani 1991).
Several studies revealed clear associations between habitat fragmentation and population
genetic structure of particularly highly mobile, long-lived and large-bodied mammal species
such as coyote and bobcat (Riley et al. 2006), roe deer (Coulon et al. 2006), bighorn sheep
(Epps et al. 2005) and grizzly bears (Proctor et al. 2002).
However, a comprehensive population genetic study has not yet been applied to free-ranging
khulans in Mongolia.
The overall objective of this study was to assess the genetic variability as well as the level of
gene flow and population differentiation of khulan populations within the framework of
landscape genetics (Manel et al. 2003) in the three main distribution areas (Great Gobi A,
Gobi B and Small Gobi) of Equus hemionus in southern Mongolia.
The secondary goals of the study were:
• to establish a panel of 10-12 equine microsatellites, that successfully cross-amplify
polymorphic loci in khulan,
• to investigate the utility of different sample material from khulan such as faeces, bones
and tissue from skeletal remains of carcasses, or preserved museum samples as potential
source of DNA for routine noninvasive genetic monitoring of khulan,
• to develop a simple and rapid khulan species determination using speciesdiscriminating PCR-RFLP of the mitochondrial cytochrome b gene to prescreen
problematic biological specimens, if the species identity is in question,
• to design a reliable protocol for microsatellite genotyping based on the preselection of
samples via qPCR (Morin et al. 2001),
• to identify spatial genetic patterns within and among khulan populations, and
• to test for correlations of genetic discontinuities with landscape and environmental
variables (Manel et al. 2003, Scribner et al. 2005).
The successful application of geo-referenced individual multilocus genotypes can provide a
basis for large-scale monitoring of khulan population responses to anthropogenic habitat
fragmentation and sustainable management strategies.
3
Materials and Methods
2 Materials and Methods
2.1 Study site
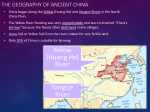
The study area in Southern Mongolia included the three SPAs: Great Gobi A, Great Gobi B
and Small Gobi (Fig. 1). The size of each study site is as follows: Gobi A - 9,000 km2, Gobi B
- 44,000 km2 and Small Gobi – 18,000 km2. Together they encompass 71,000 km² of the
potential E. hemionus distribution range in southern Mongolia. From west to east, the three
study areas are separated by the following geographic distances: GB-GA ~ 500 km, GA-GS ~
1200 km.
N= 19
N= 18
N= 43
Fig. 1: Map of the three study areas (SPAs: Great Gobi A, Great Gobi B, Small Gobi) with the corresponding
sample size and the distribution range of E. hemionus in Mongolia (Kaczensky et al. 2003)
The climate of the Gobi region is strongly continental and arid, characterized by extreme
temperatures of -35°C in winter and +45°C in summer. Precipitation averages 100mm/year
with 70 days of snow cover (Zhirnov and Ilyinsky 1986). The study area is predisposed to
large environmental fluctuations and catastrophic events that can cause large fluctuations in
wildlife and livestock population numbers (Reading et al. 2001).
Habitat conditions range from true desert areas with almost no anthropogenic use in the Gobi
A (Von Wehrden et al. in prep.), through desert-steppe areas with moderate livestock grazing
pressure in the Gobi B (Kaczensky et al. 2003, Zhirnov and Ilyinsky 1986), to desert-steppe
areas that are heavily overstocked and impacted by mining activities in parts of the Small
Gobi (Kaczensky et al. 2006).
4
Materials and Methods
2.2 Study species
E. hemionus belongs to the family of Equidae. The Gobi khulan is a grazer specialized on
monocotyledons. They prefer desert and mountain steppes and oases as year-round habitat. E.
hemionus form stable, non-territorial families and all-male groups. The life expectancy of a
free-ranging khulan is less than 12-14 years with the highest mortality rates between age
classes 4-6 years. This corresponds to the age where both sexes are at the beginning of their
reproductive period (Feh et al. 2001). Khulans have large home range sizes from 10,747 km²
to 43,105 km², as indicated by radio-telemetry data in the southeast Gobi (Kaczensky et al.
2006).
2.3 Sampling and sample preservation
A total of 80 khulan samples were collected by P. Kaczensky and members of the
International Takhi Group (ITG) in southern Mongolia during 2002 and 2005. The number of
individuals sampled in each SPA was as follows: 19 in Gobi B, 18 in Gobi A and 43 in Small
Gobi. Different kinds of biological specimens from free-ranging khulans served as source for
noninvasive genetic studies. Faecal samples (N=15) were collected from defecation sites.
Bones (N= 14) and tissue (N=51) were obtained from skeletal remains of carcasses in the
field. GPS data from each sample location were recorded. Two different sample preservation
methods were applied. Fresh faecal pellets were stored in 90% ethanol. Old faecal samples,
tissue and bones were preserved dry, packed in plastic bags and stored at -20°C prior to DNA
extraction.
2.4 Laboratory procedures
2.4.1
DNA extraction
Commercially available DNA extraction kits (QIAGEN and MACHEREY-NAGEL) were
used with some modifications to prepare highly pure genomic DNA from different kinds of
biological specimens, such as (i) deep-frozen faecal pellets, (ii) 90% ethanol preserved faecal
pellets (iii) bones and (iv) dried tissue and tendon samples.
5
Materials and Methods
(i) Frozen faecal pellets
Genomic DNA was extracted from faeces using the QIAamp® Stool Mini Kit (QIAGEN
GmbH, Germany) according to the manufacturer’s instructions but with the following
modifications:
400 mg were scraped with a razor blade from the outermost layer of each frozen faecal pellet
and incubated at room temperature in ASL buffer for 10 minutes (Wehausen et al. 2004).
Each sample was centrifuged at full speed to pellet faecal particles. 1.4 ml of the supernatant
was equally portioned on two 2 ml tubes: 350 µl 5 M NaCl and half an InhibitEx tablet were
added. Following the supplier’s instructions, ten samples and two negative controls were
processed simultaneously. Both processed solutions of each faecal sample were united and
loaded on a QIAmp spin column for DNA purification. After washing, each sample column
was centrifuged at full speed for 1 minute and additionally dried at room temperature for 10
minutes to volatilize ethanol residues. DNA from frozen faecal pellets was recovered in 100
µl of elution buffer, aliquoted and stored at -20 °C.
(ii) 90% ethanol preserved faecal pellets
Ethanol preserved faecal samples were processed like the frozen faecal pellets but with an
extra initial step for DNA extraction. After centrifugation at full speed the ethanol supernatant
was discarded and 400 mg of the remaining faecal pellet was dried at 50°C to remove ethanol
residues. DNA was finally recovered in 100 µl of BE buffer.
(iii) Bones
A layer of 1 mm was removed from the surface of the bone samples by grinding with a
drilling machine. Thus, contamination from previous handling was reduced. A drill machine
was used with low revolution speed to harvest 5 g of fine bone powder from different
locations of each bone sample. Genomic DNA from powdered bones was isolated with a
NucleoSpin Tissue Kit (MACHEREY-NAGEL GmbH & Co. KG, Germany) by using, with
some modifications, the tissue-isolation protocol provided by the manufacturer. 1 g of the
powdered bone samples was processed with the double volume of each kit reagent. The prelysis step was elongated. The bone samples were incubated at 56°C in a shaking incubator for
10 hours. Before loading the spin columns, all insoluble particles were separated by
centrifugation at full speed. The clear supernatant was transferred to a new tube and processed
according the protocol. Finally, DNA was eluted with 65 µl preheated buffer BE and stored at
–20 °C.
6
Materials and Methods
(iv) Dried tissue and tendon samples
DNA was isolated with NucleoSpin Tissue Kit (MACHEREY-NAGEL GmbH & Co. KG,
Germany) using the standard tissue-isolation protocol. DNA was recovered in 85 µl of BE
buffer.
2.4.2
Species identification
A polymerase chain reaction (PCR) coupled with a restriction fragment length polymorphism
(RFLP) analysis of the mitochondrial cytochrome b (cyt b) gene was used for discriminatory
determination of Mongolian khulan (Equus hemionus).
(i) DNA amplification
The equid-specific primer panel (Forward (CytB 1L): 5`-CTAATTAAAATCATCAATC-3`
and Reverse (CytB 2H): 5`-AAAAGTAGGATGATTCCAAT-3`) described by Orlando et al.
(2003) targets a 335-bp-long DNA fragment of the cyt b gene from perissodactylas´s mtDNA.
Amplifications were carried out in a total volume of 25 µl, containing 2 µl template DNA, 0.2
µM of each primer (CytB 2H/ CytB 1L), 0.2 mM dNTPs, 1x-PCR buffer (10x BD buffer: pH
9.4-9.5 800mM Tris-HCl, 200 mM (NH4)2SO4; Solis BioDyne Inc., Estland), 3 mM MgCl2
(Solis BioDyne Inc., Estland), 0.1 µg bovine serum albumin (BSA, Fermentas Inc.) and 1 U
®
of Taq-Polymerase (FIREPol , Solis BioDyne Inc., Estland). The PCR profile on an
Eppendorf PCR Mastergradient thermal cycler were as follows: 94°C for 3 min for
denaturation, 35 cycles of amplification (94°C for 30 s, 50°C for 30 s, 72°C for 30 s) and final
extension at 72°C for 10 minutes. PCR products were examined by electrophoresis through a
1.8% ethidium bromide stained agarose gel.
Template DNA of the Mongolian horse (Equus caballus) and the Mongolian khulan (Equus
hemionus), extracted from different biological specimens such as (i) bones, (ii) tissue, (iii)
ETOH preserved faecal pellets and (iv) deep-frozen faecal pellets were analysed.
(ii) Sequence analysis and identification of restriction sites
The amplified 335 bp long PCR products from Equus caballus and Equus hemionus were
purified (QIAquick Gel Extraction Kit) and sequenced (Sequiserve GmbH Germany). Both
sequences and the primer pair (CytB 1L, CytB 2H) were then subjected to an internetaccessible BLAST search (http://www.ncbi.nlm.nih.gov/BLAST/). Sequence information of
the cyt b gene from two additional Mongolian equids (i.e. donkey (Equus asinus) GenBank
Accession No. X97337 and Przewalski horse (Equus przewalskii) GenBank Accession No.
DQ223534) were verified. These sequences were obtained from the National Centre of
7
Materials and Methods
Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/entrez/query.
fcgi?db= Nucleotide). All equid mitochondrial cyt b sequences were aligned using MEGA3
(Kumar et al. 2004) (Fig. 2). Species-specific restriction sites of the 335 bp equid cyt b
sequences were identified with the program NEBcutter Version 2.0 (Vincze et al. 2003).
The restriction enzymes AatII (target sequence: GACGT↓C) and PagI (target sequence:
T↓CATGA) were selected by the following criteria:
Both REs produce easily distinguishable differences in RFLP banding profiles.
AatII restriction site is discriminatory for Equus hemionus (+) versus the other
Mongolian equids (-).
PagI cuts all tested equid sequences and thus failure of restriction, e.g. through
inhibitors, can be excluded.
AatII plus PagI are compatible for double digestion.
Restriction fragment patterns were expected to produce the following RFLP pattern:
Mongolian khulan (Equus hemionus) (59/131/145 bp),
Mongolian horse (Equus caballus) (59/276 bp),
Donkey (Equus asinus) (59/276 bp) and,
Przewalski horse (Equus przewalskii) (59/276 bp).
(iii) Endonuclease digestion and RFLP analysis
To test whether RFLP analysis is really diagnostic for species identification, three samples
from Equus caballus and twelve Equus hemionus from different geographical regions in
Mongolia (Small Gobi, Gobi A, Gobi B) were analysed by RFLP.
Restriction enzyme incubation with AatII plus PagI were performed in 15µl double digestion
volumes according to the manufacturer`s instruction (Fermentas). 10 µl of the PCR product
was digested with 1 U PagI (Fermentas), 1 U AatII (Fermentas), 1x restriction buffer green
(Fermentas) and 0.1 µg BSA for 1½ h at 37°C. The digested PCR products were separated on
a 1.8% ethidiumbromide stained agarose gel and visualized by ultraviolet irradiation. For size
reference, a pUC19 DNA/ MspI (HpaII) marker (Fermentas) was used.
8
Materials and Methods
Fig. 2: Sequence alignment of a 335bp fragment of the mitochondrial cyt b gene from E. hemionus (Equ_he),
E.caballus (Equ_cab_mo), E. asinus (Equ_as), E. przewalskii (Equ_pr) and the equid specific primer pair
(CytB_1L, CytB_2H). Recognition sites of the two restriction enzymes are highlighted with coloured frames, i.e.
PagI T↓CATGA (blue) and AatII GACGT↓C (red).
2.4.3
Quantitative PCR
Quantitative PCR enables the evaluation of the quality and quantity of the DNA template.
Quantification of genomic DNA in each sample was performed with real-time PCR and
SYBR Green fluorescence detection on a Light Cycler (Roche Diagnostics GmbH) (Wittwer
et al. 1997). Optimized, specific PCR primers (F: 5´GGTCGGAGTAAACGGATTTG 3´ and
R: 5´AATGAAGGGGTCATTGATGG 3´) were designed with a primer design software
program PRIMER3 (Whitehead Institute for Biomedical Research, Cambridge, MA, USA;
http://frodo.wi.mit.edu/cgibin/primer3/primer3_www.cgi) by using the GAPDH gene from
Equus caballus (Accession no. AF097178). The 100 bp target sequence was placed in the
exon of the GAPDH gene (glyceraldehyde-3-phosphate dehydrogenase), which has
9
Materials and Methods
previously been used as housekeeping gene for equine mRNA quantitative PCR (qPCR)
studies (Leuttenegger et al. 1999).
In order to evaluate the success of cross-species amplification, the GAPDH PCR product was
sequenced (Sequiserve GmbH Germany) and pairwise sequence alignment between Equus
caballus and Equus hemionus was performed using GeneDoc software.
Quantitative PCRs were performed in 10 µl reaction volume containing 1µl DNA, 0.2 µM of
each primer, 0.1 µg BSA (Fermentas), 1x LightCycler Fast Start DNA MasterPLUS (Roche
Diagnostics GmbH, Germany: MgCl2, dNTPs, Hot Start Taq and SYBR Green). The
following cycle conditions were used: initial denaturation step at 95°C for 10 min, followed
by 45 cycles at 95°C for 15 s, annealing at 58°C for 10 s, 82°C for 3 s and elongation at 72°C
for 20 s. The amplification-associated fluorescence was detected at each cycle during PCR.
Analysis was performed using Lightcyler software and quantities were checked independently
using a standard curve and calculations in Microsoft Excel. The standard curve was created
from a standard DNA dilution series of khulan tissue quantified first by absorbance (A260) in a
spectrophotometer and then by qPCR assay. DNA amounts of standard dilutions were (in 1
µl): 10 ng, 5 ng, 1 ng, 750 pg, 250 pg, 100 pg, 75 pg, 25 pg, 10 pg, 7.5 pg.
When the known initial DNA concentrations (expressed in log, X axis) are plotted against the
corresponding cycle threshold (Ct, Y axis) obtained by qPCR, the result is a line representing
the linear correlation between the two parameters. All samples including a no-template
control and three positive controls of known DNA amounts were amplified by qPCR. DNA
concentrations for each sample were calculated from the slope and Y-intercept of the trendline
from the standard curve:
DNA amount = 10 ((Ct sample –Yint STD)/slope STD)
The slope of the standard curve can be used to determine the exponential amplification and
efficiency of the PCR reaction by the following equation:
Efficiency = [10(-1/slope)] – 1
The optimal real-time PCR efficiency of 90-100% is indicated by a slope of -3.3 (Pfaffl
2001). Melting curve analysis was performed to identify sequence-specific PCR products and
exclude primer dimers and non-specific amplicons. The shape of a melting curve is a function
of GC/AT ratio, length and sequence (Ririe 1997).
2.4.4
Microsatellite amplification
Eleven equine microsatellites were selected from an initial set of fifteen loci (Bailey et al.
2000, Swinburne et al. 2000, Chowdhary et al. 2003, Guerin et al. 2003, Krüger et al. 2005):
COR70, SGCV28, ASB23, ASB2, COR58, LEX68, COR18, UM11, COR007, LEX74,
10
Materials and Methods
COR71. These were tested for successful cross-species amplification and high polymorphism
in E. hemionus. To avoid linkage, principally microsatellite loci from different chromosomes
of Equus caballus were chosen for genotyping (Genome map of the horse:
www.thearkdb.org).
PCR conditions were optimized for Equus hemionus using the following protocol:
Amplifications were carried out in a total volume of 10 µl, containing 2-4 µl template DNA,
0.3 µM of each primer (CytB 2H/ CytB 1L), 0.2 mM dNTPs, 1x-PCR buffer (10x BD buffer:
pH 9.4-9.5 800mM Tris-HCl, 200 mM (NH4)2SO4; Solis BioDyne Inc., Estland), x mM
MgCl2 (Solis BioDyne Inc., Estland) (see Table 1), 0.1 µg bovine serum albumin (BSA,
®
Fermentas Inc.) and 1 U of Taq-Polymerase (FIREPol , Solis BioDyne Inc., Estland).
The amplification conditions on a BIOMETRA UNO II thermocycler were: initial
denaturation at 95°C for 3 min, 40 cycles of 30 s at 95°C, annealing at x°C (see Table 1) for
30 s, 30s at 72°C, final elongation at 72°C for 3 min. The forward primers were fluorescently
end-labelled with Tamra, Hex or 6-Fam for genotyping with an ABI 377 DNA sequencer.
Six loci (ASB23, ASB2, COR70, SGCV28, COR58 and LEX68) were exemplarily tested
with a template DNA standard dilution series (500, 200, 100, 50, 10 and 1 pg/µl per reaction)
to examine the marker-specific minimum DNA amount necessary for reliable genotyping.
PCRs were performed with adjusted levels of standardized DNA concentration (500, 120 and
40 pg/µl per PCR reaction). One quarter of each PCR product (including a negative control)
was run on a 1.8% ethidiumbromide stained agarose gel to check amplification success and to
estimate dilutions for multiplexing on the DNA sequencer.
11
Multiplexsystem I
12
Multiplexsystem II
Multiplexsystem III
Tamra
Hex
6 Fam
F: AAGAGTGCTCCCGTGTG
R: GACAATGCAGAACTGGGTAA
F: CTTGGGCTACAACAGGGAATA
R: CTGCTATTTCAAACACTTGGA
LEX 74
COR 71
Hex
F: TGAAAGTAGAAAGGGATGTGG
R: TCTCAGAGCAGAAGTCCCTG
UM 11
F: GTGTTGGATGAAGCGAATGA
R: GACTTGCCTGGCTTTGAGTC
6 Fam
F: AGTCTGGCAATATTGAGGATGT
R: AGCAGCTACCCTTTGAATACTG
COR 18
COR 007
6 Fam
F: AAATCCCGAGCTAAAATGTA
R: TAGGAAGATAGGATCACAAGG
LEX 68
Tamra
F: CCTTCCGTAGTTTAAGCTTCTG
R: CACAACTGAGTTCTCTGATAGG
ASB 2
6 Fam
6 Fam
F: GCAAGGATGAAGAGGGCAGC
R: CTGGTGGGTTAGATGAGAAGTC
ASB 23
F: GGGAAGGACGATGAGTGAC
R: CACCAGGCTAAGTAGCCAAG
Hex
F: CTGTGGCAGCTGTCATCTTGG
R: CCCAATTCCAGCCCAGCTTGC
SGCV 28
COR 58
6 Fam
F: CATCTGTTCCGTGGCATTA
R: TTCAGGTGTGGGTTTTGAATC
di
di
di
di
di
di
di
di
di
di
di
Fluorescent Repeat
dye
type
COR 70
Genotyping
Multiplex
Locus name Primer sequences (5´- 3´)
systems
26
24
17
20
25
11
12
15
3
7
6
174-212
151-177
157-185
144-172
241-283
136-160
190-224
162-188
133-173
151-171
257-291
58
58
58
60
60
52
60
58
60
60
60
3
3
3
3
3
3
3
3
1.5
1.5
3
Ruth et al. 1999
Achmann et al. 2001
Lear et al. 1998
Godard et al. 1997
Tallmadge et al. 1999
Reference
AF142608
AF212260
AF083450
AF195130
AF083461
Tallmadge et al. 1999
Bailey et al. 2000
Hopman et al. 1999
Meyer et al. 1997
Hopman et al. 1999
ECA001605 Coogle et al. 1999
AF108375
X93516
X93537
U90604
AF142607
Optimized
Chromosome
MgCl2 GenBank
annealing
Allele
conc. Accession
location in
size (bp) temperature
(mM) no.
Equus caballus
(°C)
Table 1: Characterization of the analysed equine microsatellite loci in Mongolian khulan (E. hemionus). Locus and groupings used in multiplex genotyping gels, primer
sequences, fluorescent dye, repeat type, chromosome location in domestic horse (E. caballus), allele size (bp), optimized annealing temperature (°C), MgCl2 in mM, GenBank
accession number and reference.
Materials and Methods
Materials and Methods
2.4.5
Contamination control
Working with noninvasive genetic sampling is similar to ancient DNA studies (e.g. Stoneking
1995). Therefore the same guidelines to avoid contamination by PCR products or
concentrated genomic DNA should be followed (Taberlet et al. 1999).
In this study, the following steps were taken to avoid contamination:
(i) Pre- and post-PCR experiments were conducted at separate locations with dedicated
instruments, reagents and filter pipette tips.
(ii) Preparation of all samples was performed with glove changes between samples. All
instruments (drill bit, razor blade) and surfaces were decontaminated during
extraction process with 90% ETOH or DNA AWAY (Molecular Bio Products).
(iii) The PCR bench and pipettes were regularly decontaminated by UV light.
(iv) Blank extractions and no-template PCR controls were included during all steps of the
experiment.
2.4.6
Genotyping criteria and analysis
Genotypes were scored on an ABI Prism 377 DNA sequencer (Applied Biosystems) with
ROX 79-362 (DeWoody et al. 2004) as an internal size standard and analyzed using the
program Genescan (Applied Biosystems) and Genotyper 2.0 (Applied Biosystems) DNA
fragment analysis software.
An automated system of multiplexing, through co-electrophoresis of multiple markers in each
lane of the gel, was established. The ability to multiplex is dependant on the differences in the
relative sizes of fragments and the number of the fluorescent primer dye labels compatible
with the gel electrophoresis system (see Table 1). Using multiplex gels for genotyping post
PCR products saves time and reduces costs.
The gels were pre-run at 3 KV for 1 hour to overcome electrophoresis artefacts (Fernando et
al. 2001). All samples were electrophoresed using an additional reference sample with a
known genotype and an internal size standard (ROX 362) to ensure consistent scoring of
genotypes across all gels. 2.4 µl of the mixed PCR product received 1.4 µl formamide
(Sigma), 0.3 µl ROX 362 and was heated to 95°C for 3 minutes, immediately cooled to < 0°C
and 4.1 µl was loaded in each lane of the 6% polyacrylamide gel.
Rigid criteria for scoring and accepting consensus genotypes were applied to minimize
potential genotyping errors (Schlötterer and Tautz 1992):
(i) False peaks, which resulted from leakages of PCR products in the neighbouring
lanes, were rejected.
13
Materials and Methods
(ii) Lanes were loaded alternately with 2 minutes of short electrophoresis run in between.
(iii) Scoring of alleles was based on the existence of characteristic microsatellite stutter
bands.
(iv) Individual peaks were scored according to the peak amplitude threshold ≥ 50.
2.5 Statistical analysis
2.5.1
Intrapopulation genetic diversity
The following microsatellite diversity indices were used: Number of private alleles (AP),
average number of alleles per locus (A), mean allelic richness per population (AR), expected
and observed heterozygosity (He, Ho). All measures were calculated with GDA version 1.1
(Lewis and Zaykin 2001). GENEPOP on the Web version 3.4 (Raymond and Rousset 1995;
www.wbiomed.curtin.edu.au/genepo/) was used to measure deviations from Hardy-Weinberg
equilibrium (HWE) and linkage disequilibrium (LD) across all pairs of loci. All probability
tests were based on Markov chain Monte Carlo simulation (MCMC) with 100,000 iterations
and 1000 burn-in steps (Guo and Thompson 1992, Raymond and Rousset 1995). Sequential
Bonferroni adjustments were used to correct for the effect of multiple tests (Rice 1989).
2.5.2
Interpopulation genetic diversity
The degree of genetic divergence among putative subpopulations was estimated as pairwise
multilocus FST (Weir and Cockerham 1984) using GENEPOP on the Web.
A hierarchical analysis of molecular variance (AMOVA) implemented in ARLEQUIN
version 3.1 (Excoffier et al. 2005) was performed to partition the total variance into
covariance components at three different hierarchical levels: (i) intra-individual differences,
(ii) inter-individual differences and (iii) inter-population differences.
2.5.3
Demographic analysis
Two different methods for the detection of a recent reduction of the effective population size
through bottlenecks were carried out: (i) the ´heterozygosity excess` test (Cornuet and Luikart
1996) implemented in the program BOTTLENECK version 1.2.02 (Piry et al. 1999) and, (ii)
the ´M ratio` analysis (Garza and Williamson 2001).
• (i) The BOTTLENECK program was used to perform a Wilcoxon sign-rank test. The
significance of heterozygote excess is tested under three different mutation models twophase model (TPM), infinite allele model (IAM) and stepwise mutation model (SMM)
with 5% multistep changes and variance of 12, following the recommendations of Piry et
14
Materials and Methods
al. (1999). Heterozygosity excess indicates a population size reduction, because allelic
diversity is reduced faster than gene diversity (Nei et al. 1975, Cornuet and Luikart 1996).
• (ii) With the ´M` ratio analysis (Garza and Williamson 2001), the mean ratio of the
number of alleles (k) to the range of allele size (r) (M = k/r) was calculated for each
population. M is expected to decrease after a population size reduction because the range
in allele size at a locus (r) decreases more slowly than the number of alleles (k) under
genetic drift. The parameters were set as follows: assumed effective population size
Ne=50, mutation rate µ= 10-4 (Weber and Wong 1993).
2.5.4
Bayesian population assignment
Three different Bayesian assignment tests were applied in order to (i) infer the population
structure, (ii) identify the population of origin of individuals, and (iii) estimate recent
migration rates.
• (i) A model-based Bayesian clustering method implemented in the program
STRUCTURE 2.1 (Pritchard et al. 2000) was used to calculate the number of
subpopulations, assuming no prior information on population origin of individuals. For
the estimation of the number of subpopulations (K), five independent runs of K= 1-4 were
carried out with a burn-in period of 50,000 steps and a chain length of 500,000. The most
probable number of populations was taken using the log-likelihood of K. Individuals were
then assigned to each subpopulation, based on the highest percentage of membership (q).
• (ii) The program GENECLASS version 2 (Piry et al. 2004) was applied to estimate the
likelihood of an individual`s multilocus genoytype to be correctly assigned to its source
population (Cornuet et al. 1999, using the ´as it is`option).
• (iii) BAYESASS+ version 1.3 (Wilson and Rannala 2002) was used to estimate rates of
recent migration among populations.
2.5.5
Spatial analysis
The khulan populations were analysed within the framework of landscape genetics (Manel et
al. 2003). The basic principle of landscape genetics is to detect potential spatial population
boundaries like (i) isolation by distance (Slatkin 1993) and (ii) genetic barriers to gene flow
(Monmonier 1973). These parameters were calculated using two different software programs:
• (i) A Mantel`s test (with 1000 permutations) between the genetic differentiation [FST / (1FST)] (Rousset 1997) and the log-transformed geographical distance tests for the presence
of an isolation by distance model (IBD) (Slatkin 1993). Geographic distances between
15
Materials and Methods
pairs of sampling sites were calculated based on the coordinates of the approximate
centre of the sampling area. IBD tests were performed using Isolation By Distance Web
Service version 3.02 software (Jensen et al. 2005; www.ibdws.sdsu.edu/).
• (ii) BARRIER version 2.2 (Manni et al. 2004) was used to identify genetic boundaries
from allele frequency spatial distributions. This program uses the Monmonier maximum
difference algorithm (Monmonier 1973) on a Delaunay triangulation approach (Brassel
and Reif 1979) in order to identify zones of abrupt genetic change between different
groups of populations. The robustness of the genetic boundaries was assessed by
bootstrap (100), implemented in the BARRIER software. Geographic locations of each
sampling area were expressed in mean latitude/longitude coordinates and genetic
distances were estimated using pairwise FST-matrices (Weir and Cockerham 1984).
16
Results
3 Results
3.1 Species identification
A simple and rapid PCR-RFLP analysis of the cytochrome b was established to pre-screen
different kinds of problematic samples for reliable identification of the Mongolian khulan
(Equus hemionus).
A 335 bp fragment of the cytb gene from two Mongolian equid species was successfully
amplified from (i) bones, (ii) tissue, (iii) ETOH preserved faecal pellets and (iv) deep-frozen
faecal pellets. To confirm species identity within the Equidae family, the equid sequences
(Equ_he, Equ_ca) and the equid specific primer pair (CytB 1L, CytB 2H) were subjected to a
BLAST search. Comparison of reference versus database nucleotide sequences resulted in
highest correspondence (95-100%) with the database sequence of E. caballus.
Multiple sequence alignment of the cyt b sequences from the four Mongolian equid species
revealed interspecies polymorphisms. Pairwise alignment of E. hemionus with E. caballus
displayed fifteen point mutations within the 335 bp sequence analysed. Applicable for
analytical purposes is the transition at the position 192 bp (C↔T), enabling the discrimination
between E. caballus and E. hemionus by RFLP analysis. As expected, the double digestion
treatment with the six-cutter restriction enzymes Pag I and Aat II resulted in different, easily
distinguishable banding patterns for the Mongolian horse (Equus caballus) (59/276 bp) and
the Mongolian khulan (Equus hemionus) (59/131/145 bp) (Fig 3). The 131 and 145 bp
fragments of E. hemionus are very close in length and therefore cannot be identified as two
bands. A narrow band pattern with the original fragment size (335bp) is still present after
double digestion due to partial digestion. The intraspecific banding patterns for the twelve
reference samples of E. hemionus were consistent throughout the geographical range sampled
(see Fig 3, Lane 6-17).
17
Results
Fig. 3: RFLP banding patterns of an amplified 335 bp fragment of the cyt b gene obtained from two different
Mongolian equid species after double digestion with restriction enzymes AatII and PagI.
Lane 1: PCR product Equus caballus, undigested (335 bp); Lanes 2-4: E. caballus, digested (59/276 bp); Lane 5:
PCR product Equus hemionus, undigested (335 bp); Lanes 6-17: E. hemionus from different geographical
regions in Mongolia derived from different biological specimens (Lanes: 6-8: tissue, Lanes: 9-11: bones, Lanes:
12-14: frozen faecal pellets, Lanes 15-17: ETOH stored faecal pellets), digested (59/131/145 bp); M: pUC19
MspI size marker.
3.2 Quantitative PCR
DNA quantification was performed with real-time PCR and SYBR Green fluorescence
detection on a Light Cycler (Roche Diagnostics GmbH) (Wittwer et al. 1997). First, the
standard curve from the dilution series was created. The resulting regression line has a slope
of -3.543, Y-intercept of 37.399 and a correlation coefficient of R²= 0.9956 (Fig 4).
36.0
34.0
y = -3.543x + 37.399
R2 = 0.9956
average Ct
32.0
30.0
28.0
26.0
24.0
22.0
20.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
log DNA concentration (pg/µl)
Fig. 4: Plot of the known initial DNA concentration (expressed in log, X-axis) and the corresponding cycle
threshold (Ct, Y-axis) obtained by qPCR using primer GAPDH.
DNA concentrations calculated according to the equation of the trend line from the standard
curve are presented in Table 2.
18
Results
Table 2: DNA concentrations were calculated from the slope and Y-intercept of the trend line from the standard
curve.
DNA amount
pg/µl
10000
5000
1000
750
250
100
75
25
10
7.5
Ct values
Log pg/µl
4.0
3.7
3.0
2.9
2.4
2.0
1.9
1.4
1.0
0.9
23.0
24.4
26.8
27.1
29.2
30.0
30.9
32.8
34.0
33.9
Calculated
DNA amount
pg/µl
11366
4606
1000
786
210
119
71
19
9
6
The real-time PCR efficiency of the standard curve was 1.915 (93%).
Melting curve analysis (MCA) was performed for all samples to identify sequence-specific
PCR products and primer dimers (Ririe 1997). Sequence-specific PCR products (100bp) from
the standard curve of the equid GAPDH gene had a melting point peak at 85°C. Primer
dimers and non-specific amplicons having lower melting temperatures (80°C) were excluded
by additional fluorescence detection at 82°C (Fig 5).
Melting point peak
85 °C
Primer dimers
Fig. 5: Melting curve analysis of the standard dilution series from equid GAPDH gene amplified via qPCR.
Distribution of DNA concentration in extracts (N=80) from E. hemionus is shown in Fig. 6.
More than 77% of all extracts exceeded 1000 pg/µl (N=62). Samples with ≤ 100 pg/µl were
rejected for microsatellite DNA amplification.
19
Results
20
18
16
No. of extracts
14
12
10
8
6
4
2
0
100 - 500
500 - 1000
1000 - 5000
5000 - 10000
10000 - 50000
> 50000
DNA categories (pg/µl)
Fig. 6: Distribution of DNA concentration ( ≥100 pg/µl) in 80 extracts from E. hemionus.
Extracts obtained from the different sample material show varying concentrations of DNA
calculated via qPCR (Fig. 7). Tissue samples constitute more than 70% of the DNA categories
≥ 1000 pg/µl. The DNA concentration of 500-1000pg/µl was calculated for extracts obtained
from bones (60%) and frozen faecal pellets (40%). The DNA category of 100-500 pg/µl
consists of extracts derived from ethanol-preserved faecal pellets (40%) and frozen faecal
pellets (55%).
100%
Biological sample material in %
90%
80%
70%
60%
50%
40%
30%
20%
10%
0%
100 - 500
500 - 1000
1000 - 5000
5000 - 10000
10000 - 50000
> 50000
DNA categories (pg/µl)
Tissue
Bones
Frozen faecal pellets
Ethanol preserved faecal pellets
Fig. 7: Categorization of DNA concentrations calculated via qPCR for extracts obtained from different
biological materials like tissue, bones, frozen faecal pellets and ethanol-preserved faecal pellets.
20
Results
3.3 Microsatellite amplification and reliable genotyping
Six loci (ASB23, ASB2, COR70, SGCV28, COR58 and LEX68) were exemplarily tested
with a template DNA standard dilution series (500, 200, 100, 50, 10 and 1 pg/µl per reaction)
to examine the marker-specific minimum DNA amount necessary for reliable genotyping. All
PCR products from the DNA dilution series were diluted 1:15 for comparative scoring on the
DNA sequencer.
A correlation between genotyping errors or failed amplification with low DNA concentrations
in the PCR reaction was detected (Fig. 8). The correct single-locus genotype derived from
extracts containing high amounts of nuclear DNA (500 pg/µl per PCR reaction) is presented
in each first line of Fig. 8. Loci ASB23-6Fam and COR58-6Fam show erroneous genotypes
with allelic dropout at DNA concentrations of ≤ 50 pg/µl per reaction. Loci COR70-6Fam,
SGCV28-Hex and LEX68-6Fam show missing data at critical DNA concentrations of ≤
10pg/µl per reaction. Correct genotypes for the complete six-fold DNA dilution series were
scored for locus ASB2-tamra.
Results showed that a minimum DNA amount of 40 pg/µl per reaction was required to
reliably determine genotypes for the panel of six loci presented here.
Thus, PCRs from 80 extracts of varying DNA amounts (≥100 pg/µl) were performed with
adjusted levels of standardized DNA concentration (500, 120 and 40 pg/µl per PCR reaction).
21
Results
Allelic
dropout
Allelic
dropout
No size data
No size data
No size data
Fig. 8: Genotyping electropherogram patterns of the 6-fold DNA dilutions series amplified with the
microsatellite loci ASB2-tamra, ASB23-6Fam, COR58-6Fam, COR70-6Fam, LEX68-6Fam and SGCV28-Hex.
Genotyping errors or missing data correlated with low DNA concentrations in the PCR reaction are highlighted.
The horizontal scale represents the number of bp. The vertical scale on the right expresses the number of
fluorescence units. The Y-axis on the left shows the DNA concentrations in pg/µl per PCR reaction.
22
Results
3.4 Statistical analysis
3.4.1
Intrapopulation genetic diversity
An average number of 9.4 alleles have been observed for the eleven microsatellite loci applied
in this study (see Table 3). The average number of alleles per locus was 13.27.
Allelic variation, expressed by the average number of alleles per locus (A) and allelic richness
(AR), varied between populations. Maximum values were found in the Gobi B population (A=
9.55; AR= 9). The lowest observed values for allelic diversity (A = 8.27; AR =8.1) were found
in the Gobi A population.
Expected (HE) and observed (HO) heterozygosity were HE≥ 0.82 and HO≥ 0.70 for each
population. The number of individuals per population was positively correlated with the mean
number of alleles per locus (A), but not with allelic richness (AR), expected (HE) and observed
(HO) heterozygosity. Thus, the genetic parameters are not biased by differences in sample size
(N).
A total of 14 private alleles (AP) were detected (Gobi B= 8; Gobi A= 4 and Small Gobi= 2).
Table 3: Microsatellite diversity indices of khulan (Equus hemionus) populations in Mongolia. Sample size (N),
average number of alleles per locus (A), mean allelic richness per population (AR), number of private alleles
(AP), expected (HE) and observed (HO) heterozygosity, results of Hardy-Weinberg probability test for deviation
from expected Hardy-Weinberg proportions (PHW), test of heterozygosity excess (HE) using Wilcoxon sign-rank
test based on infinite allele model (IAM), two-phased model (TPM) and stepwise mutation model (SMM) and M
ratio (M) analysis.
Population
N
A
AR
AP
HE
HO
PHW
HE(IAM/TPM/SMM)
´M`ratio
GB
19
9.55
9
8
0.84
0.77
n.s.
y/n/n
0.727
GA
18
8.27
8.1
4
0.83
0.70
n.s.
y/n/n
0.713
GS
43
10.36
8.2
2
0.82
0.70
n.s.
y/n/n
0.765
26.67
9.39
8.43
4.67
0.83
0.72
average
n.s.: not significant (p≥ 0.05)
Linkage disequilibrium
The test for genotypic disequilibrium for each pair of the eleven microsatellite loci over all
populations gave one significant value (P < 0.05) for 55 comparisons (3 significant values are
expected by chance at the 5% level). After undertaking the Bonferroni correction for multiple
tests, none of the combinations remained significant at the experimental level (P< 0.0009).
When each population was tested separately, a linkage equilibrium between all pairs of loci
was generally observed with only few exceptions: two significant values for the Gobi B
population, one for the Gobi A population and two for the Small Gobi population. Different
23
Results
loci were involved in these cases. Generally, this test implies that the genotypes of the loci
used in this study segregated independently.
Hardy-Weinberg equilibrium
After the Bonferroni correction, the probability test using the Markov chain method based on
the ´exact HW test` of Haldane (1954) for each locus in each population showed only three
significant deviations: population Gobi A at locus COR 18 and population Small Gobi at loci
COR007 and LEX74. These deviations are not systematic, occurring at different loci and two
different populations.
3.4.2
Interpopulation genetic diversity
The highest FST values were observed between the populations Gobi B and Small Gobi. The
lowest FST values were observed between Gobi A and Small Gobi. The overall level of FST
values was low (see Table 4). Wright (1978) identified the problem of interpreting FST values
as an absolute value based on highly polymorphic loci and proposed that a FST < 0.05 still
could indicate a considerable population differentiation.
Table 4: Matrix of pairwise of FST values (Weir and Cockerham 1984) between the khulan populations in
Mongolia.
Population
GB
GA
GB
0,000
GA
0.0088
0.000
GS
0.0191
0.0068
GS
0.000
The AMOVA analysis of hierarchical gene diversity revealed that 88% of the variance is
explained by individual variation, 10.9% by variation among individuals within populations
and 1.1% by variation among populations (see Fig 9).
AMOVA
"One group- all populations"
1.1%
10.9%
Among populations
Among individuals within
populations
within individuals
88.0%
Fig. 9: AMOVA analysis conducted for three populations in one group.
24
Results
Subdivision of the three populations into two groups (K=2) defined by the model-based
Bayesian clustering method in STRUCTURE 2.1 (Pritchard et al. 2000) produced slightly
different results from AMOVA. Three possible two-group combinations were formed and
hierarchical gene diversity was compared among them:
(i) Gobi A and Small Gobi versus Gobi B
(ii) Gobi B and Small Gobi versus Gobi A
(iii) Gobi B and Gobi A versus Small Gobi
The changes of the covariance components between the three combinations are presented in
Table 5. The results of the combination (i) differ from (ii) and (iii), especially regarding the
variation among groups (1.18) which is three times higher than among populations within
groups (0.43) (see Table 5).
Table 5: AMOVA analysis performed for three group-combinations: (i) Gobi A and Small Gobi versus Gobi B,
(ii) Gobi B and Small Gobi versus Gobi A, (iii) Gobi B and Gobi A versus Small Gobi.
Percentage of variation Percentage of variation Percentage of variation
Gobi A and Small Gobi Gobi B and Small Gobi Gobi A and Gobi B
versus
versus
versus
Source of variation
Gobi B
Gobi A
Small Gobi
Among groups
1.18
-1.48
0.51
Among
populations
0.43
2.01
0.72
within groups
Among individuals
10.82
10.94
10.86
within populations
Within individuals
87.57
88.53
87.9
3.4.3
Demographic analysis
The Wilcoxon sign rank test (p< 0.05) did not detect a significant excess of heterozygosity in
the studied populations according to the two-phase model (TPM) and the stepwise mutation
model (SMM) (see Table 3). The TPM is generally expected to best reflect the mutational
process of microsatellite loci (recommended by Piry et al. 1999). Based on the infinite allele
model (IAM), all populations exhibited a significant heterozygosity excess after the
Bonferroni correction. Therefore, there is some indication of a severe reduction of effective
population sizes of all khulan populations. But this cannot be demonstrated using the
BOTTLENECK program (Cornuet and.Luikart 1996).
25
Results
Similarly, the M ratio analysis revealed that none of the populations has gone through a recent
reduction in population size. All M values were above the critical threshold value of 0.68
(Garza and William 2001) (see Table 3).
3.4.4
Bayesian population assignment
(i) Detecting cluster number in STRUCTURE
The model choice criterion implemented in STRUCTURE to detect the true number of
subpopulations (K) is an estimate of the posterior probability of the data for a given K, Pr
(X|Y) (Pritchard et al. 2000). The true number of populations (K) is identified by using the
maximum value of the mean likelihood [mean Ln(K)] returned by structure. The Bayesian
clustering model clearly indicated the presence of substructure in this sample of khulans. The
probability of data [Ln Pr(X|Y)] was maximum for K= 2 (see Fig. 10). This shows that the
three putative populations (Gobi A, B and Small Gobi) cluster into two subpopulations.
-3450
Mean Ln (K)
-3500
-3550
-3600
-3650
-3700
1
2
3
4
K
Fig. 10: Bayesian clustering results of STRUCTURE for the detection of the true number of subpopulations (K):
Plot of the estimated Mean Ln(K) over 5 runs versus the number of populations (K).
Samples were placed into the respective subpopulation based upon the highest percentage of
membership (q). The proportion of membership of each pre-defined population (3 putative
populations: Gobi A, Gobi B, Small Gobi) in each of the K= 2 clusters is shown in Table 6.
Results indicate that the samples from Gobi B and Small Gobi both cluster separately (70%).
Samples from Gobi A are equally divided into both clusters.
26
Results
Table 6: Bayesian clustering results of STRUCTURE for the assignment of individuals to each subpopulation: K
represents the number of subpopulations, with values in bold indicating the most likely values for K. For K= 2,
the average proportion of membership (q) and sample size per subpopulation cluster (in parentheses) are
presented.
Subpopulation clusters
K
Mean Ln(K)
1
-3593
2
-3532
3
-3567
4
-3678
1
2
0.692 (19)
Gobi B
0.546 (18)
Gobi A
0.71 (43)
Small Gobi
(ii) Identification of the population of origin of individuals in GENECLASS
The assignment of the individual`s multilocus genotype based on the Bayesian method
(option ´as it is`) implemented in GENECLASS version 2 (Cornuet at al. 1999) revealed that
possible source populations (Gobi B and Small Gobi) had high probability values for the
correct assignments to their origin (85% and 82%, respectively) (see I and III in Fig 11).
Probability of assigned individuals belonging to khulan population Gobi B
6%
9%
Gobi B
Gobi A
Small Gobi
85%
IA
Probability of assigned individuals belonging to khulan population Gobi A
Probability of assigned individuals belonging to khulan population Small
Gobi
18%
5%
30%
Gobi B
Gobi A
Small Gobi
Gobi B
Gobi A
Small Gobi
82%
52%
B
II
13%
C III
Fig. 11: Results of the assignment test (GENECLASS, Cornuet et al. 1999): Circular charts represent each
individual`s mean probability of belonging to its source population and to another reference population.
27
Results
The individual`s assignment of population Gobi A was much lower (52%). Some 48% of the
Gobi A population exhibited genotypes of the possible source populations, Gobi B and Small
Gobi. This result corresponds to the geographic location of the Gobi A population in the
middle of Gobi B and Small Gobi.
In contrast, only a small percentage (6%) of the Gobi B population exhibited genotypes from
the neighbouring Gobi A population. Some 9 % of the Gobi B population had genotypes from
the population Small Gobi - the population with the highest geographical distance (~1200km)
to Gobi B.
(iii) Bayesian inference of recent migration rates with BAYESASS+
Current migration rates among populations were estimated using the program BAYESASS+
version 1.3 (Wilson and Rannala 2002). Migration occurred mainly either between Gobi A
and Small Gobi (29%) (see II Fig 12) and between Gobi B and Small Gobi (26%) (see I Fig.
12). The migration pattern is predominantly unidirectional. Gene flow is directed from the
eastern Small Gobi population into the western Gobi A and Gobi B populations. There is
nearly no migration into Small Gobi (2%) (see III Fig 12).
Migration rates into khulan subpopulation Gobi B
4%
26%
Gobi B
Small Gobi
Gobi A
70%
IA
Migration rates into khulan subpopulation Gobi A
Migration rates into khulan subpopulation Small Gobi
1%
2%
1%
29%
Gobi B
Small Gobi
Gobi A
Gobi B
Small Gobi
Gobi A
69%
98%
II
B
III
C
Fig. 12: The means (averaged over posterior probabilities) of migration rates between khulan subpopulations as
detected by BAYESASS.
28
Results
3.4.5
Spatial analysis
(i) ´Isolation by distance` model
An “isolation by distance” model, defined as the significant positive correlation between
genetic and geographical distances, was not observed for the three khulan populations in the
Mantel analysis (r²= 0.0115; p> 0.05) (Fig 13).
Fig. 13: Mantel test on isolation by distance between khulan populations. Pairwise genetic distances [FST/(1FST)] (Rousset 1997) (Y-axis) are plotted against log-transformed geographic distances (geographical centre of
each sampling area) (X-axis).
(ii) Genetic boundaries
Monmonier`s maximum difference algorithm identified two barriers (I and II in Fig. 14)
showing a constant decrease from higher to lower genetic distances. The main genetic
boundary (I) separates the population of Gobi B from Gobi A with a high bootstrap value
(89%). The second barrier (II) splits the Gobi A population from Small Gobi (11%).
Gobi B
A
I
(89%)
B
II
(11%)
Gobi A
Small Gobi
Fig. 14: Detection of genetic boundaries (red lines) to gene flow between the three khulan populations in
Mongolia using the Monmonier algorithm (Monmonier 1973). Green dots indicate the geographical centre of the
populations, and blue lines show the connections of localities based on the Delauney triangulation (Brassel and
Reif 1979). The bootstrap values are shown in brackets next to the corresponding barrier.
29
Discussion
4 Discussion
4.1
Species identification
Rapid and reliable species identification from problematic biological sources (e.g.: bones,
meat, feces, blood, hair) is used in forensic science, food science and in ecological studies.
Three different techniques suitable for species identification have been developed so far:
PCR amplification of short and long interspersed elements (SINEs, LINEs) for
species-specific detection and quantification of livestock animal DNA (Walker et al.
2003).
PCR-RFLP analysis of the mitochondrial cytB (Meyer et al. 1999).
Sequencing of the mitochondrial cytb gene and a subsequent basic local alignment
search tool (BLAST) search (Brodmann et al. 2001).
Kocher et al. (1989) showed that the highly conserved regions of the mitochondrial cytb gene
are suitable for species-level identification in vertebrates and thus is the most widely applied
target gene for phylogenetic studies. Mitochondrial DNA is especially appropriate when
dealing with degraded DNA extracted from field-collected samples. Advantageous in this
record are the following mtDNA characteristics:
each diploid cell contains a high number of copies,
mtDNA is free of heterocygosity because of maternal inheritance,
mtDNA is variable enough to allow differentiation between closely related species.
In this study the relatively short length of PCR product (335 bp) and the specificity of the
equid primers contribute to the amplification success of the mitochondrial cytb.
The RFLP analysis is performed according to the double digest method of the manufacturer
(Fermentas). The RFLP setup was designed to match two criteria: (i) to produce speciesspecific, easy distinguishable banding patterns and (ii) to verify failure of restriction, e.g.
through inhibitory compounds.
Degraded DNA from various biological samples such as tissue, bones, faecal pellets proved to
be a valuable source for the identification of the study species E. hemionus. The sampling
strategy of this population genetic study on E.hemionus exclusively relies on non-invasive
collected genetic samples. Thus, the established RFLP analysis can be used to pre-screen
samples, when the species identity is in question.
30
Discussion
Indeed, PCR-RFLP analysis has been demonstrated to be an essential tool for reliable khulan
species determination, especially for the sampling strategy applied in this study. The
following facts support the application of RFLP analysis for this study:
Different Mongolian equid species co-occur in same habitat.
Identification of the species of origin from poached carcasses, meat and blood is
impossible if morphological evidence from hair, skin, tail or bone is not available.
All Mongolian equids produce optically similar pellet material.
DNA from all Mongolian equids is supposed to successfully amplify the same
microsatellite loci like E.hemionus. This is due to the fact, that the microsatellite
markers were originally developed in E. caballus and cross-amplified in E. hemionus.
Some studies revealed shortcomings of RFLP analysis of cytb for species identification.
Ambiguous RFLP banding patterns may be the result. Two determining factors were
identified: (i) the coamplification of nuclear cytb-pseudogenes (Burgener and Hübner 1998)
or (ii) intraspecies polymorphisms which occur in a restriction site by chance thus affecting
successful restriction (Wolf et al. 1999).
The RFLP method was optimised for this study to circumvent these shortcomings. First,
primers were selected to specifically amplify the mitochondrial cytb, thereby avoiding
possible co-amplification of nuclear pseudo-cytb genes. Second, the primer pair exclusively
targets DNA of species from the family Equidae. Thus non-target DNA is expected not to
amplify. Third, potential intraspecies sequence polymorphisms within the species E. hemionus
were examined in more detail by subjecting twelve individuals from different geographical
regions in Mongolia to RFLP analysis. Intraspecific banding patterns were consistent
throughout the geographical range sampled. This result confirms that either no intraspecies
polymorphisms within the 335 bp fragment of the the cytb gene occur in E.hemionus, or that
possible mutations do not affect the restriction site of the species discriminatory restriction
enzyme Aat II.
The unambiguous identification of E. hemionus via RFLP analysis might be hampered by
undetected intraspecies DNA sequence polymorphism. To fully explore the level of
intraspecific polymorphism, more individuals of E. hemionus should be sequenced and
screened for intraspecies mutations. If intraspecies polymorphisms affect the enzyme
restriction site by chance, an alternative battery of restriction enzymes has to be selected.
31
Discussion
4.2 Quantitative PCR and reliable genotyping
Quantitative PCR and SYBR Green fluorescence detection applied in this study enabled the
evaluation of the quality and quantity of the DNA template.
Other studies that have applied qPCR to noninvasively collected samples like faeces from
chimpanzee and mountain gorilla (Nsubuga et al. 2004) and feathers from capercaillie
(Segelbacher 2002) pointed out three important facts:
(i) Genotyping errors can lead to misinterpretation of genotypes (Taberlet et al. 1999)
and thus bias the inferred biological results (Creel et al. 1999).
(ii) Genotyping errors are negatively correlated with the amplifiable template DNA
amount (Morin et al. 2001).
(iii) There is an unpredictable variation of DNA quality and quantity among extracts
(Morin et al. 2001).
Thus, many studies investigated the influence of multiple variables on the DNA yield.
Determining factors are:
• Species–specific variability (Taberlet and Luikart 1999).
• Differences in diet, digestive systems and living conditions of the study species
(Murphy et al. 2003; Nsubuga et al. 2004).
• Differences in the setup of sample collection (Roeder et al. 2004), preservation
(Frantzen et al. 1998), DNA extraction (Flagstad et al. 1999) and microsatellite
amplification (Waits and Paetkau 2005).
• Locus-specific variability based on size (Buchan et al. 2005) and repeat type
(Bradley et al. 2000).
• Differences in fluorescence detection systems of qPCR: DNA binding agents
(SYBR Green: Wittwer et al. 1997) versus hydrolysis probes (5´exonuclease assay:
Livak et al. 1995).
Based on these facts, it was considered too time-consuming and cost-intensive to fully explore
the true factors affecting the DNA quality and quantity in a comprehensive khulan population
genetic study.
Thus, the preselection of samples via qPCR (Morin et al. 2001) proves to be a powerful tool
contributing to a beneficial cost-value ratio of genetic analyses by minimizing genotyping
errors. For this study an optimized real-time PCR setup based on SYBR Green fluorescence
detection was established.
32
Discussion
First, we designed primers amplifying a short 100bp fragment located in the exon of the
GAPDH gene from E. caballus. Thus, we improved species-specific amplification of
potentially degraded genomic DNA fragments retrieved from non-invasive samples
(Leutenegger 1999). A standard curve with an optimal real-time PCR efficiency of 93%
(Pfaffl 2001) was created from a 10-fold DNA standard dilution series. DNA concentrations
for each sample were calculated from the equation of the standard trendline. Additionally,
melting curve analysis (MCA) was performed and sequence-specific PCR products were
identified for all samples (Ririe 1997).
Furthermore, six loci were exemplarily tested with a template DNA standard dilution series to
analyse the locus-specific minimum DNA amount necessary for reliable genotyping.
Consequently, all extracts with a minimum DNA amount of ≥ 100pg/µl proved to be a
valuable source of template DNA for reliable genotyping the eleven equid microsatellites.
4.3 Statistical analysis
The results of our study provided good support for the hypothesis stated in the introduction as
follows:
Habitat fragmentation and potential population isolation have already led to a
detectable genetic differentiation among khulan populations in southern Mongolia.
Thus, we can draw the following two conclusions:
(i) Human disturbance of khulan habitat due to poaching and increased livestock
competition for forage and water represent a barrier to gene flow between
local khulan populations leading to a population differentiation.
(ii) The observed fragmentation of populations does not yet affect the genetic
diversity within khulan populations.
4.3.1 Intrapopulation genetic diversity
Our results indicate that all Mongolian khulan populations have a relatively high level of
overall microsatellite diversity with an average of 9.39 alleles per locus and a mean HE = 0.83
across all populations. The amount of heterozygosity examined in this study is consistent with
previous studies on other equids using the corresponding microsatellite panel (Krüger et al.
2005). We found no evidence for reduced genetic diversity within the analysed khulan
populations as reported for other highly mobile animal species suffering from habitat
33
Discussion
fragmentation [Ovis canadensis nelsoni (Epps et al. 2005) and Puma concolor (McRae et al.
2005)].
4.3.2 Demographic analysis
One reason may be that isolation due to human disturbance did not last long enough to cause a
reduced genetic diversity within fragmented populations.
Thus, demographic analysis was performed with the two most promising methods for
detecting recent bottlenecks – the ´M`ratio analysis (Garza and William 2001) and the
´heterozygosity excess`, under a two-phase mutation model (Cornuet and Luikart 1996). The
combination of both software programs was used to identify different possible genetic
scenarios (Williamson-Natesan 2005):
• ´M ratio` was the method most likely to correctly identify a bottleneck when a
bottleneck lasted several generations, the population had made a demographic
recovery, and mutation rates were high or pre-bottleneck population sizes were
large.
• The ´heterozygosity excess` test was most likely to correctly identify a bottleneck
when a bottleneck was more recent and less severe and when mutation rates were
low or pre-bottleneck population sizes were small.
Both analyses revealed a lack of severe isolation effects (see Table 3).
4.3.3 Interpopulation genetic diversity
Our analyses indicate clearly that khulan in Mongolia are not a panmictic population.
A population differentiation with at least two genetic units (K=2; STRUCTURE see Fig. 10)
were inferred, exhibiting a very low level of differentiation (pairwise FST< 0.05) (see Table
4). In this context recommendations of Wright (1978) are important, as he proposed that a
FST< 0.05 could indicate a considerable population differentiation.
Indeed, the analysis of genetic variation (AMOVA) between the three khulan populations
revealed an apparent fragmented population structure. Even though 88% of the genetic
variance is explained by within individual variation, the genetic variance explained by
variation between population Gobi B and the other population unit (Gobi A and Small Gobi)
is three times higher than the one given by variation among populations Gobi A and Small
Gobi (see (i) Table 5).
34
Discussion
The population Gobi B clusters separately from Gobi A and Small Gobi. However
microsatellite data of HE = 0.84, AR=9 and AP= 8 in the population Gobi B suggest that this
site is not genetically isolated (Table 3). Especially the high genetic diversity of the
segregated population Gobi B leads to the assumption of considerable gene flow between
Gobi B and neighbouring (un-analysed) or further distant khulan populations in the backcountry.
4.3.4 Bayesian population assignment
The three Bayesian population assignment tests applied in this study revealed the
identification of (i) population substructure with STRUCTURE (Pritchard et al. 2000), (ii) the
population of origin of individuals with GENECLASS (Piry and Cornuet 1999) and (iii)
recent migration rates with BAYESASS (Wilson and Rannala 2002).
The underlying population substructure was K=2, with population Gobi B clustering
separately as confirmed by Bayesian clustering in STRUCTURE (see Fig. 10 and Table 6).
GENECLASS results indicated that the populations Gobi B and Small Gobi showed highest
probability values (> 80%) for the correct assignment to their origin (I and III Fig.11). In
contrast, population Gobi A showed a high admixture of multilocus genotypes (48%) and
therefore a high gene flow between populations. We can assume that populations Gobi B and
Small Gobi mainly constitute the two population clusters.
BAYESASS revealed medium levels of gene flow from population Small Gobi into
populations Gobi A (29%) and Gobi B (26%) (see I and II in Fig. 12). The Bayesian approach
applied in the program BAYESASS might be sensitive to varying population sample sizes
used in this study (Gobi B=19, Gobi A=18, Small Gobi 43).
Consequently, the high genetic diversity of the segregated population Gobi B can be
explained by considerable gene flow between Small Gobi and Gobi B during former times.
This overall trend is indicated by the results of both statistical programs, GENECLASS and
BAYESASS.
4.3.5 Spatial analysis
Tools from landscape genetics were applied to test for correlations of genetic discontinuities
with landscape and environmental variables (Manel et al. 2003, Scribner et al. 2005).
The isolation by distance model (Slatkin 1993) revealed the absence of a correlation between
geographical and genetic distance at the local scale (Mantel test, Fig. 13). The lack of
35
Discussion
isolation by distance could be explained by the high dispersal rates of khulan, a conclusion
confirmed by the movements from radio-tagged khulan (Kaczensky 2006).
Consequently, it suggests that other factors potentially caused the population differentiation.
The Monmonier`s maximum difference algorithm identified two barriers to gene flow (I and
II in Fig. 14). The main genetic boundary separates the populations Gobi B from Gobi A.
The spatial genetic structure is shaped by effects of anthropogenic habitat fragmentation.
Human disturbance is no absolute and impermeable barrier to gene flow as indicated by
migration rates between Gobi B and Small Gobi. Anthropogenic barriers act as a moderator of
gene flow because of a high resistance to khulan movements, and hence their cumulative
effect has led to the differentiation of two genetic units.
Comparative studies on other animal species [Ovis canadensis nelsoni (Epps et al. 2005) and
Gulo gulo (Cegelski et al. 2003)] showed that anthropogenic barriers constitute a severe threat
to the persistence of naturally fragmented populations. We can assume that this case is also
true for khulan in Southern Mongolia. Natural barriers like the Altai mountains and
anthropogenic barriers like fenced borders and railways are already reported to block khulan
movements to other suitable habitat in Mongolia (Kaczensky et al. 2006).
36
References
5 References
Achmann R, Huber T, Wallner B, Dovc P, Muller M, Brem G (2001) Base substitutions in the
sequences flanking microsatellite markers HMS3 and ASB2 interfere with parentage
testing in the Lipizzan horse. Animal Genetics, 32, 52.
Bailey E, Skow L, Bernoco D et al. (2000) Equine dinucleotide repeat loci LEX071 through
LEX078. Animal Genetics, 31, 286–287.
Bonin A, Bellemain E, Bronken Eidesen P, et al. (2004) How to track and assess genotyping
errors in population genetics studies. Molecular Ecology, 13, 3261-3273.
Bradley BJ, Vigilant L (2002) False alleles derived from microbial DNA pose a potential
source of error in microsatellite genotyping of DNA from faeces. Molecular Ecology
Notes, 2, 602-605.
Brassel KE, Reif D (1979) A procedure to generate Thiessen polygons. Geographical
Analysis,11,3289–303
Brodmann PD, Nicholas G, Schaltenbrand P, Ilg EC (2001) Identifying unknown game
species: experience with nucleotide sequencing of the mitochondrial cytochrome b
gene and a subsequent basic local alignment search tool search. European Food
Research and Technology, 212, 491-496.
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics.
Molecular Ecology, 13, 3601-3608.
Buchan JC, Archie EA, Van Horn RC, Moss CJ, Alberts SC (2005) Locus effects and sources
of error in noninvasive genotyping. Molecular Ecology Notes, 5, 680-683.
Cassens I, Tiedemann R, Suchentrunk F, Hartl GB (2000) Mitochondrial DNAvariation in the
European otter (Lutra lutra) and
the use of spatial autocorrelation analysis in
conservation. Journal of Heredity, 91, 31– 35
Cegelski CC, Waits LP, Anderson NJ (2003) Assessing population structure and gene flow in
Montana wolverines (Gulo gulo) using assignment-based approaches. Molecular
Ecology, 12, 2907–2918.
Chowdhary BP, Raudsepp T, Kata SR et al. (2003) The First-Generation Whole-Genome
Radiation Hybrid Map in the Horse Identifies Conserved Segments in Human and
Mouse Genomes. Genome Research, 13, 742-751.
Coogle L, Bailey E (1999) Equine dinucleotide repeat loci LEX064 through LEX070.
Molecular Genetics Markers. Animal Genetics, 30, 71-72.
Cornuet JM, Luikart G (1996) Description and Power Analysis of Two Tests for Detecting
Recent Population Bottlenecks From Allele Frequency Data. Genetics, 144, 20012014.
37
References
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New Methods Employing
Multilocus Genotypes to Select or Exclude Populations as Origins of Individuals.
Genetics, 153, 1989-2000.
Coulon A, Guillot G, Cosson JFM et al. (2006) Genetic structure is influenced by landscape
features: empirical evidence from a roe deer population. Molecular Ecology, 15, 16691679.
Creel S, Spong G, Creel NM (1999) Interspecific competition and the population biology of
extinction-prone carnivores. In: Carnivore Conservation (eds Gittleman JL, Funk SM,
Macdonald D, Wayne RK), Conservation Biology 5, Cambridge University Press,
675.
DeWoody JA, Schupp J, Kenefic L (2004) Universal method for producing
ROX-labeled
size standards suitable for automated genotyping. BioTechniques, 37, 348-352.
Duncan P (1992) Zebras, asses, and horses: an Action plan for the conservation of wild
equids. IUCN, Gland Switzerland.
Epps CW, Palsbøll PJ, Wehausen JD et al. (2005) Highways block gene flow and cause a
rapid decline in genetic diversity of desert bighorn sheep. Ecology Letters, 8, 1029–
1038.
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): An integrated software
package for population genetics data analysis. Evolutionary Bioinformatics Online, 1,
47-50.
Feh C, Munkhtuya B, Enkhbold S, Sukhbaatar T (2001) Ecology and social structure of the
Gobi khulan Equus hemionus subsp. in the Gobi B National Park, Mongolia.
Biological Conservation, 101, 51-61.
Feh C, Shah N, Rowen M, Reading R, Goyal SP (2002) Status and Action Plan for the Asiatic
Wild Ass (Equus hemionus). IUCN/SSC Equid Specialist Group, Gland, Switzerland
and Cambridge, UK.
Fernando P, Pfrender ME, Encalada SE, Lande R (2000) Mitochondrial DNA variation,
phylogeography and population structure of the Asian elephant. Heredity, 84, 362–
372.
Fernando P, Evans BJ, Morales JC, Melnick DJ (2001) Electrophoresis artefacts - a
previously unrecognized cause of error in microsatellite analysis. Molecular Ecology
Notes, 1, 325–328.
Flagstad Ø, Røed K, Stacy JE, Jakobsen KS (1999) Reliable non-invasive genotyping based
on excremental PCR of nuclear DNA purified with a magnetic bead protocol.
Molecular Ecology, 8, 879-883.
Frankel OH, Soule ME (1981) Conservation and Evolution. Cambridge University Press,
366.
38
References
Frantzen MA, Silk JB, Ferguson JW, Wayne RK, Kohn MH (1998) Empirical evaluation of
preservation methods for faecal DNA. Molecular Ecology, 7, 1423-1428.
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified
concept for defining conservation units. Molecular Ecology, 10, 2741–2752.
Garnier JN, Bruford MW, Goossens B (2001) Mating system and reproductive skew in the
black rhinoceros. Molecular Ecology, 10, 2031–2041.
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from
microsatellite loci. Molecular Ecology,10, 305–318.
Godard S, Vaiman D, Oustry A et al. (1997) Characterization, genetic and physical mapping
analysis of 36 horse plasmid and cosmid-derived microsatellites. Mammalian Genome,
8, 745-750.
Goldstein DB, Schlötterer C (1999) Microsatellites: Evolution and Applications. Oxford
University Press, Oxford, 352.
Goossens B, Waits LP, Taberlet P (1998) Plucked hair samples as a source of DNA:
reliability of dinucleotide microsatellite genotyping. Molecular Ecology, 7, 12371241.
Guérin G, Bailey E, Bernoco D et al. (2003) The second generation of the International
Equine Gene Mapping Workshop half-sibling linkage map. Animal Genetics, 34, 161168.
Guo SW, Thompson EA (1992) Performing the Exact Test of Hardy-Weinberg Proportion for
Multiple Alleles. Biometrics, 48, 361-372.
Haldane JBS (1954) An exact test for randomness of mating. Journal of Genetics, 52, 631635.
Hanski, I. (1998) Metapopulation dynamics. Nature, 396, 41–49
Hardy C, Casane D, Vigne JD (2005) Ancient DNA from Bronze Age bones of European
rabbit (Oryctolagus cuniculus). Cellular and Molecular Life Sciences, 50, 564-570.
Hausknecht R, Gula R, Pirga B, Kuehn R (in prep. 2007) Urine — a source for noninvasive
genetic monitoring in wildlife. Molecular Ecology Notes.
Hedrick PW (2005) Gene flow and genetic restoration: The Florida panther as a case study.
Conservation Biology, 9, 996-1007.
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genetics,
6, 13.
Kaczensky P, Walzer C (2003) Przewalski horse, wolves and khulans in Mongolia. Report
ITG, international takhi group.
39
References
Kaczensky P, Sheehy DP, Walzer C et al. (2006) Impacts of Well Rehabilitation and Human
Intrusion on Khulan (Wild Ass) and Other Threatened Species in the Gobi Desert.
International Center for the Advancement of Pastoral Systems, the World Bank,
Netherlands-Mongolia Trust Fund for the Environment.
Kocher TD, Thomas WK, Meyer A (1989) Dynamics of mitochondrial DNA evolution in
animals: amplification and sequencing with conserved primers. Proceedings of the
National Academy of Sciences of the United States of America, 86, 6196-6200.
Kohn MH, York EC, Kamradt DA, Haught G, Sauvajot RM, Wayne RK (1999) Estimating
population size by genotyping faeces. Proceedings of the Royal Society B: Biological
Sciences, 266, 657.
Krüger K, Gaillard C, Stranzinger G, Rieder S (2005) Phylogenetic analysis and species
allocation of individual equids using microsatellite data. Journal of Animal Breeding
and Genetics, 122, 78–86.
Kumar S, Tamura K, Nei M (2004) MEGA3: Integrated software for Molecular Evolutionary
Genetics Analysis and sequence alignment. Briefings in Bioinformatics, 5, 150-163.
Lacy RC (1987) Loss of genetic diversity from managed populations: interacting effects of
drift, mutation, immigration, selection, and population subdivision. Conservation
biology, 1, 143-158.
Lande R (1998) Demographic stochasticity and Allee effect on a scale with isotropic noise.
Oikos, 83, 353-358.
Lear TL, Adams MH, Sullivan ND, McDowell KJ, Bailey E (1998) Assignment of the horse
progesterone receptor (PGR) and estrogen receptor (ESR1) genes to horse
chromosomes 7 and 31, respectively, by in situ hybridization. Cytogenetics and Cell
Genetics, 82, 110-11.
Leutenegger CM, von Rechenberg B, Huder JB et al. (1999) Quantitative Real-Time PCR for
Equine Cytokine mRNA in Nondecalcified Bone Tissue Embedded in Methyl
Methacrylate. Calcified Tissue International, 65, 378-383.
Lewis PO, Zaykin D (2001) Genetic Data Analysis: Computer program for the analysis of
allelic data. Version 1.0: http://lewis.eeb.uconn.edu/lewishome/software.html.
Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K (1995) Oligonucleotides with fluorescent
dyes at opposite ends provide a quenched probe system useful for detecting PCR
product and nucleic acid hybridization. PCR. Methods and Applications, 14, 357-362.
M. Burgener, P. Hübner (1998) Mitochondrial DNA enrichment for species identification and
evolutionary analysis. Zeitschrift für Lebensmitteluntersuchung und -Forschung A,
207, 261-263.
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining
landscape ecology and population genetics. Trends in Ecology and Evolution, 18, 189197.
40
References
Manni F (2004) Geographic Patterns of (Genetic, Morphologic, Linguistic) Variation: How
Barriers Can Be Detected by Using Monmonier's Algorithm. Human Biology, 76, 173190.
MCrae BH, Beier P, Dewald LE, Huynh LY, Keim P (2005) Habitat barriers limit gene flow
and illuminate historical events in a wide-ranging carnivore, the American puma.
Molecular Ecology, 14, 1965–1977.
Meyer R, Hofelein C, Luthy J, Candrian U (1995) Polymerase chain reaction-restriction
fragment length polymorphism analysis: a simple method for species identification in
food. Journal of AOAC International, 78, 1542-51.
Meyer AH, Valberg SJ, Hillers KR, Schweitzer JK, Mickelson JR (1997) Sixteen new
polymorphic equine microsatellites. Molecular Genetic Markers. Animal Genetics, 28,
69-70.
Miller CR, Joyce P, Waits LP (2002) Assessing allelic dropout and genotype reliability using
maximum likelihood. Genetics, 160, 357-366.
Monmonier MS (1973) Maximum-Difference Barriers:
regionalization Method. Geographical Analysis, 3.
An Alternative numerical
Morin PA, Woodruff DS (1996) Noninvasive genotyping for vertebrate conservation. In:
Molecular Genetic Approaches in Conservation (eds Smith TB, Wayne RK), pp. 298–
313. Oxford University Press, New York.
Morin PA, Chambers KE, Boesch C, Vigilant L (2001) Quantitative polymerase chain
reaction analysis of DNA from noninvasive samples for accurate microsatellite
genotyping of wild chimpanzees (Pan troglodytes verus). Molecular Ecology, 10,
1835-1844.
Murphy MA, Waits LP, Kendall KC (2003) The influence of diet on faecal DNA
amplification and sex identification in brown bears (Ursus arctos). Molecular Ecology,
12, 2261–2265.
Navidi W, Arnheim N, Waterman MS (1992) A multiple-tubes approach for accurate
genotyping of very small DNA samples by using PCR: statistical considerations.
American Journal of Human Genetics, 50, 347–359.
Nei M, Maruyama T, Chakraborty R (1975) The Bottleneck Effect and Genetic Variability in
Populations. Evolution, 29, 1-10.
Nsubuga AM, Robbins MM, Roeder AD, et al. (2004) Factors affecting the amount of
genomic DNA extracted from ape faeces and the identification of an improved sample
storage method. Molecular Ecology, 13, 2089-2094.
Orlando L, Eisenmann V, Reynier F, Sondaar P, Hänni C (2003) Morphological Convergence
in Hippidion and Equus (Amerhippus) South American Equids Elucidated by Ancient
DNA Analysis. Journal of Molecular Evolution, 57, 29-40.
41
References
Paetkau D (2003) An empirical exploration of data quality in DNA-based population
inventories. Molecular Ecology, 12, 1375-1387.
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT–
PCR. Nucleic Acids Research, 29, 45.
Pierntey SB, Maccoll ADC, Bacon PJ, Dallas JF (1998) Local genetic structure in red grouse
(Lagopus lagopus scoticus): evidence from microsatellite DNA markers. Molecular
Ecology, 7, 1645.
Piglucci M, Barbujani G (1991) Geographical patterns of gene frequencies in Italian
populations of Ornithogalum montanum (Liliaceae). Genetical Research, 58, 95–104.
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting
recent reductions in the effective size using allele frequency data. Journal of Heredity,
90, 502-503.
Piry S, Alapetite A, Cornuet JM et al. (2004) GENECLASS2: A Software for Genetic
Assignment and First-Generation Migrant Detection. Journal of Heredity, 95, 536–
539.
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes,
consequences and solutions. Nature Reviews Genetics, 6, 847-859.
Pritchard JK, Stephens M, Donnelly P (2000) Inference of Population Structure Using
Multilocus Genotype Data. Genetics, 155, 945-959.
Proctor M, McLellan BN, Strobeck C (2003) Population fragmentation of grizzly bears in
southeastern British Columbia, Canada. Ursus, 15, 153-160.
Raymond M, Rousset F (1995) GENEPOP (Version 1.2): Population Genetics Software for
Exact Tests and Ecumenicism. Journal of Heredity, 86, 248-249.
Reading RP, Mix HM, Lhagvasuren B (2001) Status and distribution of khulan (Equus
hemionus) in Mongolia. Journal of Zoology, 254, 381-389.
Rice WR (1989) Analyzing Tables of Statistical Tests. Evolution, 43, 223-225.
Riley SPD, Pollinger JP, Sauvajot RM et al. (2006) A southern California freeway is a
physical and social barrier to gene flow in carnivores. Molecular Ecology, 15,
1733–1741.
Ririe KM, Rasmussen RP, Wittwer CT (1997) Product Differentiation by Analysis of DNA
Melting Curves during the Polymerase Chain Reaction. Analytical Biochemistry, 245,
154–160.
Roeder AD, Archer FI, Poinar HN, Morin PA (2004) A novel method for collection and
preservation of faeces for genetic studies. Molecular Ecology Notes, 4, 761-764.
Rousset F (1997) Genetic Differentiation and Estimation of Gene Flow from FStatistics under
Isolation by Distance. Genetics, 145, 1219-1228.
42
References
Ruth LS, Hopman TJ, Schug MD et al. (1999) Equine dinucleotide repeat loci COR041COR060. Molecular Genetic Markers. Animal Genetics, 30, 320-321.
Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, Hanski I (1998) Inbreeding and
extinction in a butterfly metapopulation. Nature, 392, 491.
Schlötterer C, Tautz D (1992) Slippage synthesis of simple sequence DNA. Nucleic Acids
Research, 20, 211-215.
Scribner KT, Blanchong JA, Bruggeman DJ et al. (2005) Geographical genetics: conceptual
foundations and empirical applications of spatial genetic data in wildlife management.
Journal of Wildlife Management, 69, 1434–1453.
Segelbacher G (2002) Noninvasive genetic analysis in birds: testing reliability of feather
samples. Molecular Ecology Notes, 2, 367–369.
Slatkin M (1993) Isolation by Distance in Equilibrium and Non-Equilibrium Populations.
Evolution, 47, 264-279.
Sokal RR, Thomson BA (1998) Spatial genetic structure of human populations in Japan.
Human Biology, 70, 1–22.
Stoneking M. (1997) Ancient DNA: how do you know when you have it and what can you do
with it? American Journal of Human Genetics, 60, 1259-1262.
Swinburne J E, Lockhart L, Aldridge V et al. (2000) Characterisation of 25 new physically
mapped horse microsatellite loci: AHT24-48. Animal Genetics, 31, 237-238.
Taberlet P, Bouvet J (1992) Bear conservation genetics. Nature, 358, 197.
Taberlet P, Fumagalli L (1996) Owl pellets as a source of DNA for genetic studies of small
mammals. Molecular Ecology, 5 , 301–305.
Taberlet P, Luikart G (1999) Non-invasive genetic sampling
identification. Biological Journal of the Linnean Society, 68, 41-55.
and
individual
Taberlet P, Waits LP, Luikart G (1999) Noninvasive genetic sampling: look before you leap.
Trends Ecology and Evolution, 14, 323-327.
Tallmadge RL, Evans KG, Hopman TJ et al. (1999) Equine dinucleotide repeat loci
COR081-COR100. Molecular Genetic Markers. Animal Genetics, 30, 470.
Tallmadge RL, Hopman TJ, Schug MD et al. (1999) Equine dinucleotide repeat loci cor061cor080. Animal Genetics, 30, 462–466.
Tischendorf L, Fahrig L (2000) How should we measure landscape connectivity? Landscape
Ecology, 15, 633-641.
Valiere N, Taberlet P (2000) Urine collected in the field as a source of DNA for species and
individual identification. Molecular Ecology, 9, 2150–2152.
43
References
Valière N, Berthier P, Mouchiroud D, Pontier D (2002) Gemini: software for testing the
effects of genotyping errors and multitubes approach for individual identification.
Molecular Ecology Notes, 2, 83–86.
Valière N, Fumagalli L, Gielly L, Miquel C, Lequette B, Poulle ML, Weber JM, Arlettaz R,
Taberlet P (2003) Long-distance wolf recolonization of France and Switzerland
inferred from non-invasive genetic sampling over a period of 10 years. Animal
Conservation, 6, 83-92.
Valsecchi E (1998) Tissue boiling: a short-cut in DNA extraction for large-scale population
screenings. Molecular Ecology, 7, 1243–1245.
Vincze T, Posfai J, Roberts RJ (2003) NEBcutter: a program to cleave DNA with
restriction enzymes. Nucleic Acids Research, 31, 3688-3691.
Von Wehrden H, Wesche K (2005) Mapping the vegetation of southern Mongolian protected
areas:application of GIS and remote sensing techniques. In: Dorjsuren Ch, Dorofeyuk
NI, Gunin PD, Ecosystems of Mongolia and frontier areas of adjacent countries:
natural resources, biodiversity and ecological prospects. - Publishing House "Bembi
San", Ulaanbaatar: 232-234.
Waits LP, Paetkau D (2005) Noninvasive genetic sampling tools for wildlife biologists: a
review of applications and recommendations for accurate data collection. Journal of
Wildlife Management, 69, 1419-1433.
Walker AJ, Hughes AD, Hedges JD et al. (2004) Quantitative PCR for DNA identification
based on genome-specific interspersed repetitive elements. Genomics, 83, 518-527.
Wandeler P, Smith S, Morin PA, Pettifor RA, Funk SM (2003) Patterns of nuclear DNA
degeneration over time - a case study in historic teeth samples. Molecular Ecology, 12,
1087–1093.
Weber JL, Wong C (1993) Mutation of human short tandem repeats. Human Molecular
Genetics, 2, 1123-1128.
Wehausen J D, Ramey RR, Epps CW (2004) Experiments in DNA Extraction and PCR
Amplification from Bighorn Sheep Feces: the Importance of DNA Extraction Method.
Journal of Heredity, 95, 503–509.
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population
structure. Evolution, 38, 1358-1370.
Wiens JA (1996) Wildlife in patchy environments: metapopulations, mosaics, and
management. In: Metapopulations and Wildlife Conservation Management (ed.
McCullough D). pp.53-84. Island Press, Washington, D.C..
Wilcox A, Murphy D (1985) Conservation Strategy: The Effects of Fragmentation on
Extinction. The American Naturalist, 125, 879-887.
44
References
Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from
microsatellite loci. Conservation Genetics, 6, 551-562.
Wilson GA, Rannala B (2003) Bayesian Inference of Recent Migration Rates Using
Multilocus Genotypes. Genetics, 163, 1177–1191.
Wittwer CT, Ririe KM, Andrew RV et al. (1997) The LightCycler: a microvolume
multisample fluorimeter with rapid temperature control. Biotechniques, 22, 176-181.
Wolf C, Rentsch J, Hübner P (1999) PCR-RFLP Analysis of Mitochondrial DNA: A Reliable
Method for Species Identification. Journal of Agricultural and Food Chemistry, 47,
1350 -1355.
Worley K, Strobeck C, Arthur S et al. (2004) Population genetic structure of North
American thinhorn sheep (Ovis dalli). Molecular Ecology, 13, 2545–2556.
Wright S (1965) The Interpretation of Population Structure by F-Statistics with Special
Regard to Systems of Mating. Evolution, 19, 395-420.
Zhirnov L, Ilinski V (1986) The Great Gobi National Park, a refuge for rare animals of the
Central Asian Deserts. UNEP, Moscow.
45
Appendix I
APPENDIX I
Table A1: Results from the ´M´ratio analysis of the three populations GB, GA, GS.
GA_50
4*Ne*(mutation rate)= 0.2 %larger mutations = 0.1 mean step size = 3.5
Locus 0: # alleles = 14 range = 16 ratio = 0.875 sample size = 36
Locus 1: # alleles = 7 range = 11 ratio = 0.636364 sample size = 34
Locus 2: # alleles = 12 range = 14 ratio = 0.857143 sample size = 30
Locus 3: # alleles = 8 range = 9 ratio = 0.888889 sample size = 32
Locus 4: # alleles = 9 range = 20 ratio = 0.45 sample size = 36
Locus 5: # alleles = 7 range = 10 ratio = 0.7 sample size = 32
Locus 6: # alleles = 6 range = 10 ratio = 0.6 sample size = 36
Locus 7: # alleles = 6 range = 11 ratio = 0.545455 sample size = 36
Locus 8: # alleles = 5 range = 5 ratio = 1 sample size = 36
Locus 9: # alleles = 10 range = 14 ratio = 0.714286 sample size = 34
Locus 10: # alleles = 7 range = 12 ratio = 0.583333 sample size = 36
Average M = 0.713679
Simulating 10000 replicates, please wait...
0.05% of the time you expect a smaller ratio at equilibrium
GB_50
4*Ne*(mutation rate)= 0.2 %larger mutations = 0.1 mean step size = 3.5
Locus 0: # alleles = 14 range = 21 ratio = 0.666667 sample size = 38
Locus 1: # alleles = 9 range = 12 ratio = 0.75 sample size = 36
Locus 2: # alleles = 11 range = 13 ratio = 0.846154 sample size = 38
Locus 3: # alleles = 9 range = 13 ratio = 0.692308 sample size = 36
Locus 4: # alleles = 7 range = 12 ratio = 0.583333 sample size = 38
Locus 5: # alleles = 11 range = 18 ratio = 0.611111 sample size = 38
Locus 6: # alleles = 7 range = 8 ratio = 0.875 sample size = 38
Locus 7: # alleles = 9 range = 11 ratio = 0.818182 sample size = 38
Locus 8: # alleles = 8 range = 11 ratio = 0.727273 sample size = 38
Locus 9: # alleles = 11 range = 14 ratio = 0.785714 sample size = 38
Locus 10: # alleles = 9 range = 14 ratio = 0.642857 sample size = 38
Average M = 0.727145
Simulating 10000 replicates, please wait...
0.14% of the time you expect a smaller ratio at equilibrium
GS_50
4*Ne*(mutation rate)= 0.2 %larger mutations = 0.1 mean step size = 3.5
Locus 0: # alleles = 15 range = 17 ratio = 0.882353 sample size = 86
Locus 1: # alleles = 8 range = 12 ratio = 0.666667 sample size = 84
Locus 2: # alleles = 11 range = 13 ratio = 0.846154 sample size = 86
Locus 3: # alleles = 8 range = 9 ratio = 0.888889 sample size = 86
Locus 4: # alleles = 12 range = 22 ratio = 0.545455 sample size = 86
Locus 5: # alleles = 11 range = 12 ratio = 0.916667 sample size = 84
Locus 6: # alleles = 8 range = 12 ratio = 0.666667 sample size = 84
Locus 7: # alleles = 6 range = 7 ratio = 0.857143 sample size = 82
Locus 8: # alleles = 11 range = 15 ratio = 0.733333 sample size = 72
Locus 9: # alleles = 14 range = 20 ratio = 0.7 sample size = 86
Locus 10: # alleles = 10 range = 14 ratio = 0.714286 sample size = 86
Average M = 0.765237
Simulating 10000 replicates, please wait...
0.36% of the time you expect a smaller ratio at equilibrium
46
Appendix II
APPENDIX II
Table A2: Results from the WILCOXON TEST in BOTTLENECK.
Estimation based on 1000 replications.
Population : GB
WILCOXON TEST
Assumptions: all loci fit I.A.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.99976
Probability (one tail for H excess): 0.00049
Probability (two tails for H excess and deficiency): 0.00098
Assumptions: all loci fit T.P.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.96631
Probability (one tail for H excess): 0.04150
Probability (two tails for H excess or deficiency): 0.08301
Assumptions: all loci fit S.M.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.23242
Probability (one tail for H excess): 0.79346
Probability (two tails for H excess or deficiency): 0.46484
Population : GA
WILCOXON TEST
Assumptions: all loci fit I.A.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.99976
Probability (one tail for H excess): 0.00049
Probability (two tails for H excess and deficiency): 0.00098
Assumptions: all loci fit T.P.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.98950
Probability (one tail for H excess): 0.02686
Probability (two tails for H excess or deficiency): 0.05371
Assumptions: all loci fit S.M.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.87988
Probability (one tail for H excess): 0.13916
Probability (two tails for H excess or deficiency): 0.27832
Population : GS
WILCOXON TEST
Assumptions: all loci fit I.A.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.99951
Probability (one tail for H excess): 0.00073
Probability (two tails for H excess and deficiency): 0.00146
Assumptions: all loci fit T.P.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.83984
Probability (one tail for H excess): 0.18262
Probability (two tails for H excess or deficiency): 0.36523
Assumptions: all loci fit S.M.M., mutation-drift equilibrium.
Probability (one tail for H deficiency): 0.01050
Probability (one tail for H excess): 0.99194
Probability (two tails for H excess or deficiency): 0.02100
47
Appendix III
APPENDIX III
Table A3: Results for the ´Genotypic disequilibrium´ analysis in GenePop web version 3.4.
Number of population detected: 3
Numer of loci detected:
11
Markov chain parameters
Dememorization:
1000
Batches:
100
Iterations per batch: 1000
Pop
Locus#1 Locus#2
P-Value
------- ------- ------------------GB
ASB23
ASB2
0.03074
GB
ASB23
COR70
0.05293
GB
ASB2
COR70
0.19139
GB
ASB23
SGCV28
0.05590
GB
ASB2
SGCV28
0.43341
GB
COR70
SGCV28
0.14873
GB
ASB23
COR18
1.00000
GB
ASB2
COR18
0.27597
GB
COR70
COR18
0.42795
GB
SGCV28 COR18
1.00000
GB
ASB23
COR58
0.12365
GB
ASB2
COR58
0.12703
GB
COR70
COR58
0.13238
GB
SGCV28 COR58
0.16804
GB
COR18
COR58
1.00000
GB
ASB23
LEX68
0.11333
GB
ASB2
LEX68
0.69892
GB
COR70
LEX68
0.47099
GB
SGCV28 LEX68
0.62997
GB
COR18
LEX68
1.00000
GB
COR58
LEX68
0.32488
GB
ASB23
UM11
1.00000
GB
ASB2
UM11
0.28149
GB
COR70
UM11
1.00000
GB
SGCV28 UM11
1.00000
GB
COR18
UM11
1.00000
GB
COR58
UM11
1.00000
GB
LEX68
UM11
1.00000
GB
ASB23
COR007
0.09729
GB
ASB2
COR007
0.32054
GB
COR70
COR007
0.45784
GB
SGCV28 COR007
0.62046
GB
COR18
COR007
0.95533
GB
COR58
COR007
0.39585
GB
LEX68
COR007
0.06070
GB
UM11
COR007
0.63709
GB
ASB23
COR71
0.00582
GB
ASB2
COR71
0.07238
GB
COR70
COR71
0.13895
GB
SGCV28 COR71
0.10185
GB
COR18
COR71
1.00000
GB
COR58
COR71
0.02683
GB
LEX68
COR71
0.19789
GB
UM11
COR71
1.00000
GB
COR007 COR71
0.21082
S.E.
0.01404
0.02096
0.03197
0.02138
0.04202
0.03061
0.00000
0.02649
0.03295
0.00000
0.03112
0.02910
0.03133
0.03236
0.00000
0.02447
0.02829
0.03505
0.03511
0.00000
0.03674
0.00000
0.03558
0.00000
0.00000
0.00000
0.00000
0.00000
0.02522
0.03130
0.03742
0.03809
0.00792
0.04245
0.01467
0.03624
0.00582
0.02453
0.03264
0.02839
0.00000
0.01373
0.02929
0.00000
0.03626
48
Appendix III
GB
GB
GB
GB
GB
GB
GB
GB
GB
GB
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
GA
ASB23
LEX74
1.00000
0.00000
ASB2
LEX74
1.00000
0.00000
COR70
LEX74
1.00000
0.00000
SGCV28 LEX74
1.00000
0.00000
COR18
LEX74
1.00000
0.00000
COR58
LEX74
1.00000
0.00000
LEX68
LEX74
0.34816
0.03480
UM11
LEX74
1.00000
0.00000
COR007 LEX74
1.00000
0.00000
COR71
LEX74
0.08356
0.02603
ASB23
ASB2
1.00000
0.00000
ASB23
COR70
1.00000
0.00000
ASB2
COR70
1.00000
0.00000
ASB23 SGCV28
no information
ASB2
SGCV28
1.00000
0.00000
COR70
SGCV28
1.00000
0.00000
ASB23
COR18
1.00000
0.00000
ASB2
COR18
0.71680
0.02259
COR70
COR18
1.00000
0.00000
SGCV28 COR18
0.31400
0.03127
ASB23
COR58
no information
ASB2
COR58
0.28334
0.03170
COR70
COR58
1.00000
0.00000
SGCV28 COR58
1.00000
0.00000
COR18
COR58
0.07295
0.01363
ASB23
LEX68
1.00000
0.00000
ASB2
LEX68
0.61089
0.02900
COR70
LEX68
1.00000
0.00000
SGCV28 LEX68
0.27840
0.03306
COR18
LEX68
0.11566
0.01945
COR58
LEX68
0.37283
0.03165
ASB23
UM11
1.00000
0.00000
ASB2
UM11
1.00000
0.00000
COR70
UM11
1.00000
0.00000
SGCV28 UM11
0.39083
0.02976
COR18
UM11
1.00000
0.00000
COR58
UM11
1.00000
0.00000
LEX68
UM11
0.72579
0.02309
ASB23
COR007
0.10012
0.02074
ASB2
COR007
1.00000
0.00000
COR70
COR007
0.12210
0.01904
SGCV28 COR007
1.00000
0.00000
COR18
COR007
0.16644
0.01663
COR58
COR007
0.44822
0.02285
LEX68
COR007
0.78851
0.02120
UM11
COR007
0.51612
0.02459
ASB23
COR71
1.00000
0.00000
ASB2
COR71
0.40543
0.03429
COR70
COR71
0.04410
0.01721
SGCV28 COR71
1.00000
0.00000
COR18
COR71
0.47354
0.03209
COR58
COR71
1.00000
0.00000
LEX68
COR71
1.00000
0.00000
UM11
COR71
0.54349
0.03398
COR007 COR71
0.13681
0.02173
ASB23
LEX74
1.00000
0.00000
ASB2
LEX74
1.00000
0.00000
COR70
LEX74
1.00000
0.00000
SGCV28 LEX74
1.00000
0.00000
COR18
LEX74
0.14173
0.02298
COR58
LEX74
1.00000
0.00000
LEX68
LEX74
0.33174
0.03260
49
Appendix III
GA
GA
GA
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
GS
UM11
COR007
COR71
ASB23
ASB23
ASB2
ASB23
ASB2
COR70
ASB23
ASB2
COR70
SGCV28
ASB23
ASB2
COR70
SGCV28
COR18
ASB23
ASB2
COR70
SGCV28
COR18
COR58
ASB23
ASB2
COR70
SGCV28
COR18
COR58
LEX68
ASB23
ASB2
COR70
SGCV28
COR18
COR58
LEX68
UM11
ASB23
ASB2
COR70
SGCV28
COR18
COR58
LEX68
UM11
COR007
ASB23
ASB2
COR70
SGCV28
COR18
COR58
LEX68
UM11
COR007
COR71
LEX74
LEX74
LEX74
ASB2
COR70
COR70
SGCV28
SGCV28
SGCV28
COR18
COR18
COR18
COR18
COR58
COR58
COR58
COR58
COR58
LEX68
LEX68
LEX68
LEX68
LEX68
LEX68
UM11
UM11
UM11
UM11
UM11
UM11
UM11
COR007
COR007
COR007
COR007
COR007
COR007
COR007
COR007
COR71
COR71
COR71
COR71
COR71
COR71
COR71
COR71
COR71
LEX74
LEX74
LEX74
LEX74
LEX74
LEX74
LEX74
LEX74
LEX74
LEX74
1.00000
0.54136
0.30704
1.00000
1.00000
0.78736
1.00000
0.04736
0.28816
1.00000
0.05726
0.73117
0.92550
1.00000
0.45283
0.42648
0.18124
0.49024
0.64211
0.49135
0.83729
0.71654
0.29763
0.08677
0.53530
0.96051
0.93292
0.46681
0.97747
1.00000
0.32391
1.00000
0.67114
0.77407
0.76317
0.15182
0.48980
0.29921
0.90565
1.00000
0.59121
0.00983
0.15625
0.93249
0.23850
0.91168
0.88234
0.84562
0.26997
0.09533
0.05223
0.18917
0.22038
0.04983
0.27091
0.74333
0.28664
0.73379
0.00000
0.02913
0.03911
0.00000
0.00000
0.03359
0.00000
0.01571
0.04021
0.00000
0.01620
0.04103
0.01878
0.00000
0.04326
0.04809
0.03521
0.04584
0.04298
0.03639
0.03255
0.03040
0.03758
0.02175
0.04493
0.01100
0.02133
0.03623
0.01040
0.00000
0.03288
0.00000
0.03641
0.03542
0.03151
0.03001
0.04500
0.03519
0.02009
0.00000
0.04256
0.00699
0.03299
0.02269
0.04136
0.02523
0.02575
0.03248
0.04300
0.02243
0.02145
0.03228
0.03489
0.01780
0.03980
0.03674
0.04120
0.04038
P-value for each locus pair across all populations
(Fisher's method)
--------------------------------------------------------
50
Appendix III
Locus pair
Chi2
df
------------------------- ---ASB23 & ASB2
6.964
ASB23 & COR70
5.878
ASB2
& COR70
3.785
ASB23 & SGCV28 5.768
ASB2
& SGCV28 7.772
COR70 & SGCV28 6.300
ASB23 & COR18
0.000
ASB2
& COR18
8.961
COR70 & COR18
2.324
SGCV28 & COR18
2.472
ASB23 & COR58
4.181
ASB2
& COR58
8.233
COR70 & COR58
5.749
SGCV28 & COR58
6.983
COR18 & COR58
6.662
ASB23 & LEX68
5.241
ASB2
& LEX68
3.123
COR70 & LEX68
1.861
SGCV28 & LEX68
4.148
COR18 & LEX68
6.738
COR58 & LEX68
9.111
ASB23 & UM11
1.250
ASB2
& UM11
2.616
COR70 & UM11
0.139
SGCV28 & UM11
3.403
COR18 & UM11
0.046
COR58 & UM11
0.000
LEX68 & UM11
2.896
ASB23 & COR007 9.263
ASB2
& COR007 3.073
COR70 & COR007 6.280
SGCV28 & COR007 1.495
COR18 & COR007 7.448
COR58 & COR007 4.886
LEX68 & COR007 8.492
UM11
& COR007 2.423
ASB23 & COR71
10.293
ASB2
& COR71
8.108
COR70 & COR71
19.435
SGCV28 & COR71
8.281
COR18 & COR71
1.635
COR58 & COR71
10.103
LEX68 & COR71
3.425
UM11
& COR71
1.470
COR007 & COR71
7.427
ASB23 & LEX74
2.619
ASB2
& LEX74
4.701
COR70 & LEX74
5.904
SGCV28 & LEX74
3.330
COR18 & LEX74
6.932
COR58 & LEX74
5.998
LEX68 & LEX74
6.929
UM11
& LEX74
0.593
COR007 & LEX74
3.726
COR71 & LEX74
7.945
P-value
---------6
0.324
6
0.437
6
0.706
4
0.217
6
0.255
6
0.390
6
1.000
6
0.176
6
0.888
6
0.872
4
0.382
6
0.222
6
0.452
6
0.322
6
0.353
6
0.513
6
0.793
6
0.932
6
0.657
6
0.346
6
0.167
6
0.974
6
0.855
6
1.000
6
0.757
6
1.000
6
1.000
6
0.822
6
0.159
6
0.800
6
0.393
6
0.960
6
0.281
6
0.559
6
0.204
6
0.877
6
0.113
6
0.230
6
0.003
6
0.218
6
0.950
6
0.120
6
0.754
6
0.961
6
0.283
6
0.855
6
0.583
6
0.434
6
0.766
6
0.327
6
0.423
6
0.327
6
0.997
6
0.714
6
0.242
Normal ending.
51
Appendix V
APPENDIX IV
Table A4: Results for the Pairwise FST for population pairs implemented in Genepop web
version of 3.4.
(FST is estimated as in Weir and Cockerham 1984)
Number of samples detected:
3
Number of loci detected:
11
Code for pop names:
---- ------------1
GB
2
GA
3
GS
---------------------Estimates for each locus:
------------------------ASB23:
-------------------pop 1
2
2 0.0231
3 0.0528 0.0048
LEX68:
-------------------pop 1
2
2 0.0170
3 0.0123 0.0149
ASB2:
-------------------pop 1
2
2 0.0441
3 0.0000 0.0190
UM11:
-------------------pop 1
2
2 0.0006
3 -0.0154 0.0201
COR70:
-------------------pop 1
2
2 0.0057
3 0.0434 0.0456
COR007:
-------------------pop 1
2
2 -0.0265
3 -0.0035 -0.0068
SGCV28:
-------------------pop 1
2
2 0.0070
3 0.0183 -0.0052
COR71:
-------------------pop 1
2
2 0.0116
3 0.0160 -0.0085
COR18:
-------------------pop 1
2
2 -0.0160
3 0.0138 -0.0010
LEX74:
-------------------pop 1
2
2 -0.0009
3 0.0524 -0.0038
COR58:
-------------------pop 1
2
2 0.0203
3 0.0068 -0.0065
Estimates for all loci:
------------------------pop 1
2
2 0.0088
3 0.0191 0.0068
Normal ending
52
Appendix V
APPENDIX V
Table V: Results from the AMOVA analysis with different group combinations in Arlequin
3.1.
No. of Groups = 1
StructureName = "New Edited Structure"
NbGroups = 1
IndividualLevel = 0
DistMatLabel = ""
Group={"GA""GB""GS")- one group= all populations
Distance method: No. of different alleles (FST)
Reference: Weir, B.S. and Cockerham, C.C. 1984.
Excoffier, L., Smouse, P., and Quattro, J. 1992.
Weir, B. S., 1996.
---------------------------------------------------------------------Source of
Sum of
Variance
Percentage
variation
d.f.
squares components
of variation
---------------------------------------------------------------------Among
populations
2
14.419
0.04999 Va
1.14
Among
individuals
within
populations
77
369.131
0.47508 Vb
10.87
Within
individuals 80
307.500
3.84375 Vc
87.98
---------------------------------------------------------------------Total
159
691.050
4.36882
----------------------------------------------------------------------
No. of Groups = 2
StructureName = "New Edited Structure"
NbGroups = 2
IndividualLevel = 0
DistMatLabel = ""
Group={"GA""GS")
Group="GB"
---------------------------------------------------------------------Source of
Sum of
Variance
Percentage
variation
d.f.
squares components
of variation
---------------------------------------------------------------------Among
groups
1
8.659
0.05161 Va
1.18
Among
populations
within
groups
1
5.760
0.01904 Vb
0.43
Among
individuals
within
53
Appendix V
populations 77
369.131
0.47508 Vc
10.82
Within
individuals 80
307.500
3.84375 Vd
87.57
---------------------------------------------------------------------Total
159
691.050
4.38948
---------------------------------------------------------------------No. of Groups = 2
StructureName = "New Edited Structure"
NbGroups = 2
IndividualLevel = 0
DistMatLabel = ""
Group="GA"
Group={"GB""GS" }
---------------------------------------------------------------------Source of
Sum of
Variance
Percentage
variation
d.f.
squares components
of variation
---------------------------------------------------------------------Among
groups
1
5.037
-0.06420 Va
-1.48
Among
populations
within
groups
1
9.383
0.08706 Vb
2.01
Among
individuals
within
populations 77
369.131
0.47508 Vc
10.94
Within
individuals 80
307.500
3.84375 Vd
88.53
---------------------------------------------------------------------Total
159
691.050
4.34168
---------------------------------------------------------------------No. of Groups = 2
StructureName = "New Edited Structure"
NbGroups = 2
IndividualLevel = 0
DistMatLabel = ""
Group={"GS"}
Group={"GB""GA"}
---------------------------------------------------------------------Source of
Sum of
Variance
Percentage
variation
d.f.
squares components
of variation
---------------------------------------------------------------------Among
groups
1
8.462
0.02250 Va
0.51
Among
populations
within
groups
1
Among
individuals
within
populations
77
5.957
369.131
0.03147 Vb
0.47508 Vc
0.72
10.86
54
Appendix V
Within
individuals 80
307.500
3.84375 Vd
87.90
---------------------------------------------------------------------Total
159
691.050
4.37280
----------------------------------------------------------------------
55
Appendix VI
APPENDIX VI
Table A6: Results of HE and HO per locus and per population calculated with GDA version
1.1.
Data matrix has 3 populations, 11 loci, and 80 individuals
Descriptive statistics for locus: ASB23
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
14
14
2 GA
18
1
14
14
3 GS
43
1
15
15
---------- ---------- ---------- ---------- ---------Mean 26.666667 1.000000 14.333333 14.333333
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.921764 0.894737 0.030111
2 GA 0.920635 1.000000 -0.088968
3 GS 0.899590 0.906977 -0.008310
---------- ---------- ---------- ---------Mean 0.913996 0.933905 -0.022195
Descriptive statistics for locus: ASB2
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
18
1
9
9
2 GA
17
1
7
7
3 GS
42
1
8
8
---------- ---------- ---------- ---------- ---------Mean 25.666667 1.000000 8.000000
8.000000
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.831746 0.777778 0.066667
2 GA 0.795009 0.882353 -0.113689
3 GS 0.775674 0.571429 0.265672
---------- ---------- ---------- ---------Mean 0.800810 0.743853 0.073945
Descriptive statistics for locus: COR70
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
11
11
2 GA
15
1
12
12
3 GS
43
1
11
11
---------- ---------- ---------- ---------- ---------Mean 25.666667 1.000000 11.333333 11.333333
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.883357 0.842105 0.047934
2 GA 0.912644 0.866667 0.052083
3 GS 0.837483 0.790698 0.056492
---------- ---------- ---------- ----------
56
Appendix VI
Mean
0.877828
0.833157
0.052136
Descriptive statistics for locus: SGCV28
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
18
1
9
9
2 GA
16
1
8
8
3 GS
43
1
8
8
---------- ---------- ---------- ---------- ---------Mean 25.666667 1.000000 8.333333
8.333333
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.880952 0.722222 0.184502
2 GA 0.846774 0.687500 0.193154
3 GS 0.818605 0.744186 0.091892
---------- ---------- ---------- ---------Mean 0.848777 0.717969 0.156905
Descriptive statistics for locus: COR18
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
7
7
2 GA
18
1
9
9
3 GS
43
1
12
12
---------- ---------- ---------- ---------- ---------Mean 26.666667 1.000000 9.333333
9.333333
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.729730 0.578947 0.211155
2 GA 0.776190 0.388889 0.506224
3 GS 0.793981 0.697674 0.122563
---------- ---------- ---------- ---------Mean 0.766634 0.555170 0.279157
Descriptive statistics for locus: COR58
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
11
11
2 GA
16
1
7
7
3 GS
42
1
11
11
---------- ---------- ---------- ---------- ---------Mean 25.666667 1.000000 9.666667
9.666667
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.899004 0.894737 0.004878
2 GA 0.826613 0.500000 0.402985
3 GS 0.868904 0.714286 0.179727
---------- ---------- ---------- ---------Mean 0.864840 0.703008 0.190324
Descriptive statistics for locus: LEX68
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
7
7
2 GA
18
1
6
6
57
Appendix VI
3 GS
42
1
8
8
---------- ---------- ---------- ---------- ---------Mean 26.333333 1.000000 7.000000
7.000000
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.833570 0.894737 -0.075571
2 GA 0.814286 0.833333 -0.024096
3 GS 0.766208 0.738095 0.037121
---------- ---------- ---------- ---------Mean 0.804688 0.822055 -0.021822
Descriptive statistics for locus: UM11
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
9
9
2 GA
18
1
6
6
3 GS
41
1
6
6
---------- ---------- ---------- ---------- ---------Mean 26.000000 1.000000 7.000000
7.000000
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.823613 0.684211 0.173145
2 GA 0.750794 0.666667 0.114967
3 GS 0.785306 0.731707 0.069046
---------- ---------- ---------- ---------Mean 0.786571 0.694195 0.119562
Descriptive statistics for locus: COR007
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
8
8
2 GA
18
1
5
5
3 GS
36
1
11
11
---------- ---------- ---------- ---------- ---------Mean 24.333333 1.000000 8.000000
8.000000
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.742532 0.684211 0.080550
2 GA 0.717460 0.500000 0.309255
3 GS 0.756260 0.638889 0.157068
---------- ---------- ---------- ---------Mean 0.738751 0.607700 0.180638
Descriptive statistics for locus: COR71
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
11
11
2 GA
17
1
10
10
3 GS
43
1
14
14
---------- ---------- ---------- ---------- ---------Mean 26.333333 1.000000 11.666667 11.666667
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.871977 0.789474 0.096990
2 GA 0.887701 0.764706 0.142268
58
Appendix VI
3 GS 0.872230 0.674419 0.228870
---------- ---------- ---------- ---------Mean 0.877303 0.742866 0.156734
Descriptive statistics for locus: LEX74
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB
19
1
9
9
2 GA
18
1
7
7
3 GS
43
1
10
10
---------- ---------- ---------- ---------- ---------Mean 26.666667 1.000000 8.666667
8.666667
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.859175 0.684211 0.208122
2 GA 0.826984 0.611111 0.266667
3 GS 0.798085 0.511628 0.361658
---------- ---------- ---------- ---------Mean 0.828081 0.602317 0.277737
Descriptive statistics (by population):
Population
n
P
A
Ap
---------- ---------- ---------- ---------- ---------1 GB 18.818182 1.000000 9.545455 9.545455
2 GA 17.181818 1.000000 8.272727 8.272727
3 GS 41.909091 1.000000 10.363636 10.363636
---------- ---------- ---------- ---------- ---------Mean 25.969697 1.000000 9.393939 9.393939
Population
He
Ho
f
---------- ---------- ---------- ---------1 GB 0.843402 0.767943 0.091741
2 GA 0.825008 0.700111 0.155374
3 GS 0.815666 0.701817 0.141043
---------- ---------- ---------- ---------Mean 0.828025 0.723290 0.129150
Descriptive statistics (by locus):
Locus
n
P
A
Ap
---------- ---------- ---------- ---------- ---------ASB23 80.000000 1.000000 20.000000 20.000000
ASB2 77.000000 1.000000 11.000000 11.000000
COR70 77.000000 1.000000 16.000000 16.000000
SGCV28 77.000000 1.000000 10.000000 10.000000
COR18 80.000000 1.000000 16.000000 16.000000
COR58 77.000000 1.000000 14.000000 14.000000
LEX68 79.000000 1.000000 10.000000 10.000000
UM11 78.000000 1.000000 10.000000 10.000000
COR007 73.000000 1.000000 13.000000 13.000000
COR71 79.000000 1.000000 15.000000 15.000000
LEX74 80.000000 1.000000 11.000000 11.000000
---------- ---------- ---------- ---------- ---------All 77.909091 1.000000 13.272727 13.272727
Locus
He
Ho
f
---------- ---------- ---------- ---------ASB23 0.925708 0.925000 0.000769
ASB2 0.801884 0.688312 0.142431
59
Appendix VI
COR70 0.883966 0.818182 0.074872
SGCV28 0.844071 0.727273 0.139159
COR18 0.778459 0.600000 0.230363
COR58 0.871912 0.714286 0.181756
LEX68 0.800048 0.797468 0.003245
UM11 0.788586 0.705128 0.106446
COR007 0.740671 0.616438 0.168698
COR71 0.880593 0.721519 0.181592
LEX74 0.832940 0.575000 0.311025
---------- ---------- ---------- ---------All 0.831713 0.717146 0.138519
60
Appendix VII
APPENDIX VII
Table 7: Results of the Hardy-Weinberg test per locus aand population calculated in Genepop
web version of 3.4.
Number of populations detected: 3
Number of loci detected:
11
Estimation of exact P-values
by the Markov chain method
--------------------------------------------Markov chain parameters for all tests
Dememorization
: 5000
Batches
: 500
Iterations per batch : 5000
Results by locus
====================================================
Locus: ASB23
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.7108 0.0066 +0.030 +0.054 GA
0.9856 0.0013 -0.089 -0.050 GS
0.8437 0.0045 -0.008 -0.012 All (Fisher's method) :
chi2 : 1.051538
Df : 6
Prob: 0.983583
Locus: ASB2
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.0900 0.0028 +0.067 +0.044 GA
0.4963 0.0032 -0.114 -0.057 GS
0.0316 0.0013 +0.266 +0.215 All (Fisher's method) :
chi2 : 13.124575
Df : 6
Prob: 0.041100
Locus: COR70
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.1560 0.0042 +0.048 +0.095 GA
0.5197 0.0069 +0.052 +0.067 GS
0.0807 0.0031 +0.056 +0.089 All (Fisher's method) :
61
Appendix VII
chi2 : 10.058878
Df : 6
Prob: 0.122194
Locus: SGCV28
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.0126 0.0008 +0.185 +0.260 GA
0.1438 0.0026 +0.193 +0.309 GS
0.0512 0.0013 +0.092 +0.219 All (Fisher's method) :
chi2 : 18.564795
Df : 6
Prob: 0.004965
Locus: COR18
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.0172 0.0008 +0.211 +0.141 GA
0.0000 0.0000 +0.506 +0.566 GS
0.0046 0.0008 +0.123 +0.067 All (Fisher's method) :
chi2 : Infinity
Df : 6
Prob: High. sign.
Locus: COR58
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.3398 0.0053 +0.005 +0.016 GA
0.0177 0.0008 +0.403 +0.382 GS
0.0653 0.0022 +0.180 +0.184 All (Fisher's method) :
chi2 : 15.679942
Df : 6
Prob: 0.015579
Locus: LEX68
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.0067 0.0004 -0.076 -0.001 GA
0.2185 0.0018 -0.024 +0.140 GS
0.1085 0.0025 +0.037 +0.271 All (Fisher's method) :
chi2 : 17.480860
Df : 6
62
Appendix VII
Prob: 0.007669
Locus: UM11
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.3105 0.0043 +0.173 +0.066 GA
0.0747 0.0013 +0.115 +0.244 GS
0.5773 0.0021 +0.069 +0.014 All (Fisher's method) :
chi2 : 8.625926
Df : 6
Prob: 0.195734
Locus: COR007
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.0936 0.0026 +0.081 -0.000 GA
0.0447 0.0007 +0.309 +0.293 GS
0.0009 0.0003 +0.157 +0.311 All (Fisher's method) :
chi2 : 24.958443
Df : 6
Prob: 0.000348
Locus: COR71
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.7207 0.0050 +0.097 +0.031 GA
0.0100 0.0008 +0.142 +0.085 GS
0.0059 0.0008 +0.229 +0.121 All (Fisher's method) :
chi2 : 20.122300
Df : 6
Prob: 0.002634
Locus: LEX74
------------------------------------------------------------------Fis:
-----------------------POP
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----GB
0.2158 0.0036 +0.208 +0.128 GA
0.1119 0.0018 +0.267 +0.213 GS
0.0000 0.0000 +0.362 +0.208 All (Fisher's method) :
chi2 : Infinity
Df : 6
Prob: High. sign.
63
Appendix VII
Results by population
====================================================
Pop: GB
-----------------------------------------------------------Fis:
------------------LOCUS
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----ASB23
0.7108 0.0066 +0.030 +0.054 ASB2
0.0900 0.0028 +0.067 +0.044 COR70
0.1560 0.0042 +0.048 +0.095 SGCV28
0.0126 0.0008 +0.185 +0.260 COR18
0.0172 0.0008 +0.211 +0.141 COR58
0.3398 0.0053 +0.005 +0.016 LEX68
0.0067 0.0004 -0.076 -0.001 UM11
0.3105 0.0043 +0.173 +0.066 COR007
0.0936 0.0026 +0.081 -0.000 COR71
0.7207 0.0050 +0.097 +0.031 LEX74
0.2158 0.0036 +0.208 +0.128 All (Fisher's method) :
chi2 : 49.0403
Df : 22
Prob: 0.0008
Pop: GA
-----------------------------------------------------------Fis:
------------------LOCUS
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----ASB23
0.9856 0.0013 -0.089 -0.050 ASB2
0.4963 0.0032 -0.114 -0.057 COR70
0.5197 0.0069 +0.052 +0.067 SGCV28
0.1438 0.0026 +0.193 +0.309 COR18
0.0000 0.0000 +0.506 +0.566 COR58
0.0177 0.0008 +0.403 +0.382 LEX68
0.2185 0.0018 -0.024 +0.140 UM11
0.0747 0.0013 +0.115 +0.244 COR007
0.0447 0.0007 +0.309 +0.293 COR71
0.0100 0.0008 +0.142 +0.085 LEX74
0.1119 0.0018 +0.267 +0.213 All (Fisher's method) :
chi2 : Infinity
Df : 22
Prob: High. sign.
Pop: GS
-----------------------------------------------------------Fis:
------------------LOCUS
P-val S.E W&C R&H Matr
------------------------ ------ ------ ------ ------ -----ASB23
0.8437 0.0045 -0.008 -0.012 ASB2
0.0316 0.0013 +0.266 +0.215 COR70
0.0807 0.0031 +0.056 +0.089 SGCV28
0.0512 0.0013 +0.092 +0.219 COR18
0.0046 0.0008 +0.123 +0.067 COR58
0.0653 0.0022 +0.180 +0.184 -
64
Appendix VII
LEX68
UM11
COR007
COR71
LEX74
0.1085 0.0025 +0.037 +0.271
0.5773 0.0021 +0.069 +0.014
0.0009 0.0003 +0.157 +0.311
0.0059 0.0008 +0.229 +0.121
0.0000 0.0000 +0.362 +0.208
-
All (Fisher's method) :
chi2 : Infinity
Df : 22
Prob: High. sign.
65
Acknowledgements
6 Acknowledgments
This work was conducted at the Technische Universität München, funded by the working
group ´Molecular Ecology and Conservation Genetics`. I would like to thank Petra Kaczensky
from the ITG (International takhi group) for providing samples from the Mongolian khulan. I
would also like to thank Ralph Kühn and Bernhard Gum for supervision. Many thanks to my
friends Eike Müller, Kristina Salzer and Helmut Bayerl for inspiring discussions and for their
support. I highly appreciated their friendly company during field trips in the Alps, thus we
compensated the everyday life in the lab. Furthermore, I thank my mother for her
encouragement and her financial support.
66
Eidesstattliche Erklärung
Eidesstattliche Erklärung
Ich erkläre hiermit wahrheitsgemäß, dass ich
-
die eingereichte Arbeit selbständig und ohne unerlaubte Hilfsmittel angefertigt habe,
-
nur die im Literaturverzeichnis aufgeführten Hilfsmittel benutzt und fremdes
Gedankengut als solches kenntlich gemacht habe,
-
alle Personen und Institutionen, die mich bei der Vorbereitung und Anfertigung der
Abhandlung unterstützt haben, genannt habe und
-
die Arbeit noch keiner anderen Stelle zur Prüfung vorgelegt habe
Ort, Datum
Unterschrift (Vor- und Zuname)
67