Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Therapeutic gene modulation wikipedia , lookup
Gene nomenclature wikipedia , lookup
Protein moonlighting wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Fetal origins hypothesis wikipedia , lookup
Tay–Sachs disease wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Designer baby wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Point mutation wikipedia , lookup
Microevolution wikipedia , lookup
Medical genetics wikipedia , lookup
Genome (book) wikipedia , lookup
Public health genomics wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
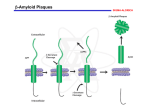
Disorders with Complex Genetics Alzheimer’s Disease Signs & Symptoms: • Memory loss for recent events • Progresses into dementia almost total memory loss • Inability to converse, loss of language ability • Affective/personality disturbance (fatuous, hostile) • Death from opportunistic infections, etc. Confirmation of Diagnosis: • Neuronal (amyloid, b amyloid, Ab amyloid) plaques • Neurofibrillary tangles • Brain Atrophy Neuronal Plaques in Alzheimer’s Disease From http://www.rnw.nl/health/html/brain.html Neurofibrillary Tangles in Alzheimer’s Disease From http://www.rnw.nl/health/html/brain.html Plaques and neurofibrillary tangles From Department of Pathology, Virginia Commonwealth University http://www.hosppract.com/genetics/9707gen.htm Brain Atrophy in AD WRONG! http://abdellab.sunderland.ac.uk/lectures/Neurodegeneration/References/Brain_Neurons_AD_Normal.html Classification: (1) FAD v SAD: Familial AD versus Sporadic AD • No complete consensus • Usually FAD = at least 1 first degree relative affected • Sometimes 2 second degree relatives (2) Early v Late Onset: • Early onset (EOAD) = usually before 65 • Early onset correlated with FAD • LOAD = late onset AD Alzheimer’s Disease, Type 1: •Several mutations in APP gene on chromosome 21 •Most common = Val717Iso •Produce abnormal beta amyloid fragment •15%-20% of early onset, familial AD •Autosomal dominant http://ghr.nlm.nih.gov/condition=alzheimerdisease http://perso.wanadoo.fr/alzheimer.lille/APP/APPmutations.html Alzheimer’s Disease, Type 3: •Mutations (> 130) in the presenilin1 gene on chromosome 14 •Most mutations lead to amino acid substitution •Overproduction of the beta amyloid fragment •30% - 70% of early onset, familial AD •Autosomal dominant Alzheimer’s Disease, Type 4: • Mutations in the presenilin2 gene on chromosome 1 • 2 alleles: Asn141Iso and Met239Val • Overproduction of the beta amyloid fragment • < 5% of early onset, familial AD (only a few families world wide) • Autosomal dominant Alzheimer’s Disease, Type 2: • Epsilon 4 (e4, AKA E4) allele of the Apolipoprotein E (ApoE) gene on chromosome 19 confers risk • Epsilon 2 (e2, AKA E2) allele of the Apolipoprotein E gene on chromosome 19 confers protection • Mechanism unclear; ApoE is a very low density lipoprotein that transports cholesterol • Most cases are late onset, familial • Susceptibility Locus Prevalence of APOE genotypes in Alzheimer’s disease (AD) and controls. Genotype: Controls AD E2/E2 1.3% 0% E2/E3 12.5% 3.4% E2/E4 4.9% 4.3% E3/E3 59.9% 38.2% E3/E4 20.7% 41.2% E4/E4 0.7% 12.9% Jarvik G, Larson EB, Goddard K, Schellenberg GD, Wijsman EM (1996) Influence of apolipoprotein E genotype on the transmission of Alzheimer disease in a community-based sample. Am J Hum Genet 58:191-200 http://www.hosppract.com/genetics/9707gen.htm Two Major Hypotheses for AD: b amyloid protein (BAP) v. tau 1. BAPtists: The accumulation of a fragment of the amyloid precursor protein or APP (the amyloid beta 42 residue fragment or Ab-42) leads to the formation of plaques that somehow kill neurons. 2. TAUists: Abnormal phosphorylation of tau proteins makes them “sticky,” leading to the break up of microtubules. The resulting loss of axonal transport causes cell death. (Recently a presenilin hypothesis has been proposed by Shen & Kelleher (2007), PNAS, 104:403-408.) Amyloid Hypothesis (it’s the plaques, dummy) 1. The amyloid precursor protein (APP) is broken down by a series of secretases (see next two slides). 2. During this process, a nonsoluble fragment of the APP protein (called Ab42) accumulates and is deposited outside the cell. 3. The nonsoluble or “sticky” nature of Ab-42 helps other protein fragments (including apoE) to gather into plaques. 4. Somehow the plaques (or possible the migration of Ab-42 outside the cell) cause neuronal death. 5. PSEN1 & PSEN2 genes subunits of g secretase. Amyloid precursor protein (APP) is membrane protein that sits in the membrane and extends outward. It is though to be important for neuronal growth, survival, and repair. From: www.niapublications.org/pubs/unraveling/01.htm Enzymes cut the APP into fragments, the most important of which for AD is called b-amyloid (beta-amyloid) or Ab. From: www.niapublications.org/pubs/unraveling/01.htm Beta-amyloid is “sticky” so the fragments cling together along with other material outside of the cell, forming the plaques seen in the AD brain. From: www.niapublications.org/pubs/unraveling/01.htm b-secretase Pathway: (not drawn to scale) APP Protein: b a g g (1) b-secretase cuts APP protein, giving: (2) g-secretase cuts this residue, giving: Ab40 Fragment Soluble Ab42 Fragment Unsoluble, aggregates into plaques or Tau Hypothesis (it’s the tangles, dummy) 1. Ordinarily, the t (tau) protein is a microtubule-associated protein that acts as a three-dimensional “railroad tie” for the microtubule. The microtubule is responsible for axonal transport. 2. Accumulation of phosphate on the tau proteins cause “paired helical filaments” or PHFs (like two ropes twisted around each other) that accumulate and lead to the neurofibrillary tangles (NFT). PHFs are the main component in NFTs. 3. Impaired axonal transport is the probable cause of cell death. 4. Focus on MAPT gene (microtubule-associated protein tau) 5. Not in favor anymore. Microtubules are like railroad tracks that transport nutrition and other molecules. Tau-proteins act as “ties” that stabilize the structure of the microtubules. In AD, tau proteins become tangled, unstabilizing the structure of the microtubule. Loss of axonal transport results in cell death. AD: The Great Unknown: What is causing the majority of AD cases? 1. Unknown Mendelian forms (probably not) 2. Unknown major loci (probably not) 3. Phenocopies 4. Multifactorial threshold a. Unidimensional b. Heterogeneity Phenocopy Environmental insult that will produce the disorder in anyone regardless of genotype • Heavy metal poisoning and ID • Amphetamine (stimulant) psychosis Head injury: possible phenocopy for ADS Testing a helmet. From: http://dailysanctuary.com/a-rare-photos-from-the-past/ Multifactorial Threshold Model • Many alleles with “low” penetrance. • Most people will have several risk alleles. • Many environmental factors. • Genetic and environmental factors add together liability •Once liability reaches a certain value (i.e., the threshold) a disease process begins. Multifactorial Threshold Model Notes: • Alleles can be common or rare • Factors can be multiplicative (taking the log makes them additive) • Central limit guarantees a normal distribution to liability • Can have more than one threshold Frequency High Unaffected Affected Low Low Medium Liability High Heterogeneity I: Mendelian/Phenocopy • Many rare alleles with high penetrance (“Mendelian” forms of the disorder). • Few will have two or more of these AD alleles. • Non familial cases due to phenocopies. Heterogeneity II: Multifactorial Several pathways to AD, each sufficient to cause the disorders Current theories: • Protein accumulation: placques & tangles • Inflammation: Unregulated activation of glia • Lipid distribution: Lipid membrane site of APP cleavage. Unidimensional vs Heterogeneity Multifactorial Threshold Model UMTF v HMTF • UMTF: different mechanisms can compensate • HMTF: other mechanisms cannot compensate E.g., In the HMTF model, if a person passes the threshold on the cholesterol dimension they will develop AD regardless of their liability on other dimensions. In the UMTF model, low liability on endocytosis can compensate for high liability on cholesterol Note Well: Mathematics allows for BOTH unidimensional and heterogeneity MFT models at the same time. From Karch & Goate (2014), Biological Psychiatry preprint From Sleegers et al. (2010) Trends in Genetics, 26, 84-94, p. 87 Alzheimers Disease http://www.ambion.com/tools/pathway/pathway.php?pathway=Alzheimer's%20Disease%20Pathway Current gene candidates for AD: • Changes too rapidly to keep track of. • Go to http://Alzgene.org for latest list DCGs: Three Models Gibson (2012) Nature Genetics 1.Infinitesimal model (CDCV) 2.Rare allele model (CDRV) 3.Broad sense heritability model Cases with no known etiology: (theoretical extremes) Mendelian/ Phenocopy Disease (Genetic) Heterogeneity Multifactorial/ Threshold CDCV Common disease/ common variant CDCV Common disease, common variant 1.Many risk alleles 2.Common (MAF > 1%) 3.Add together risk Rare Variants Common Disease, Rare Variant (CDRV_ 1.Many variants with frequencies < 1% 2.Large effect size 3.Prob(Dis | variant) is large 4. genetic heterogenetity Broad sense heritability 1.Significant dominance and epistatic variance 2.Gene-environment interaction 3. Epigenetic effects small Effect Size large Current Gospel Rare Common Allele Frequency Rare Variants: Advantages: 1. Mendelian disorders are rare 2. Selection vs alleles reducing fitness Disadvantages: 1. Inconsistent with recurrence risks 2. GWAS has found common variants Common Variants: Advantages: 1. GWAS has found common alleles Disadvantages: 1. Still have “missing” h2 2. Statistical effects vs functional effects Implications CDCV = More GWAS! CDRV = Wait till sequencing is mature! Broad h2 = ? Animal Models Human APP gene Mice gratia http://www.kidscolorpages.com/mouse.htm Human ApoE gene Human Presenilin gene Figure 1. Development of the Transgenic Mouse Model of Alzheimer's Disease. The transgene consists of the human APP gene containing a mutation causing a rare form of early-onset familial Alzheimer's disease (Val717Phe). The transgene, whose expression is driven by the platelet-derived growth factor (PDGF) promoter, is microinjected into mouse eggs and implanted in a pseudopregnant female mouse. After the progeny are screened for the presence of the transgene, they are bred and their offspring are analyzed for pathologic features characteristic of Alzheimer's disease. The brains of the transgenic PDAPP (PDGF promoter expressing amyloid precursor protein) mice have abundant -amyloid deposits (made up of the A peptide), dystrophic neurites, activated glia, and overall decreases in synaptic density. From NEJM Volume 332:1512-1513 From McGowan, Erikson & Hutton (2006), Trend in Genetics, 22: 281-289.