Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Aging brain wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Perivascular space wikipedia , lookup
Premovement neuronal activity wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Visual selective attention in dementia wikipedia , lookup
Alzheimer's disease wikipedia , lookup
Parkinson's disease wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
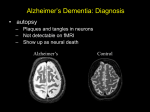
Neurodegenerative Diseases Neurodegenerative Diseases These are diseases of gray matter characterized: • by the progressive loss of neurons • with associated secondary changes in white matter tracts. Neurodegenerative Diseases • The pattern of neuronal loss is selective, affecting one or more group of neurons. • A common theme is the presence of protein aggregates resistant to degredation through the ubiquitinproteasome system. Neurodegenerative diseases • Characterized by nerve cell loss and gliosis, often in specific regions of the brain which correlate with clinical symptoms – example: Huntington diseasecaudate nucleus • Specific or non-specific inclusions are common • Gross atrophy of the affected regions may be seen— • Examples: – Diffuse atrophy in Alzheimer disease, – Caudate atrophy in Huntington disease. Neurodegenerative disorders • Degenerative diseases affecting the cerebral cortex Alzheimer disease Frontotemporal dementias Pick disease Progressive supranuclear palsy Corticobasal degeneration Vascular dementia • Degenerative diseases of basal ganglia and brain stem Parkinson disease- Akinetic Dementia with Lewy bodies Huntington disease- Hyperkinetic • Spinocerebellar degenerations Spinocerebellar ataxia Friedrich ataxia Ataxia telengiectasia • Degenerative diseases affecting motor neurons ALS Bulbospinal atrophy Spinal muscular atrophy Degenerative diseases affecting the cerebral cortex Degenerative diseases affecting the cerebral cortex • Alzheimer disease • Dementia of frontotemporal lobe type ; DFLT) • Pick disease • Other – Vascular dementia (serebrosclerosis) – Dementia with hydrocephalus (NPH). Dementia • Neuropathologically – Alzheimer disease 65% – Vascular disease 15-20% – Dementia with Lewy bodies 10% – Others: • Dementia of frontal lobe type – Pick disease (<2%) Alzheimer's Disease • Degenerative; • The major cause of dementia in the elderly; • Main pathologic change is generalized atrophy of brain with neurofibrillary tangles, senile plaques and amyloid angiopathy in many parts of the brain. • Impairment of recent memory • Aphasia (naming), apraxia (motor), agnosia (object), executive functioning • Progressive over time • 47% of people over 85 years of age are affected • Familial cases with a defined inheritance pattern account for only 5 to 10% of Alzheimer's disease. – Familial cases tend to have an earlier age at onset. – Genetic defects in familial cases have been identified on chromosomes 21, 19, 14, 12 and 1. • Regardless of the cause, the diagnosis of AD is made clinically by the finding of progressive memory loss with increasing inability to participate in activities of daily living. – Late in the course of the disease, affected persons are not able to recognize family members and may not know who they are. • The definitive diagnosis is made pathologically by examination of the brain at autopsy. • Grossly, there is cerebral atrophy, mainly in frontal, temporal, and parietal regions. • As a consequence, there is ex vacuo ventricular dilation. Alzheimer's disease leads to cerebral atrophy. The external surface of the brain with widened sulci and narrowed gyri, mostly over the frontal and parietal regions, gross. Alzheimer's disease : the characteristic hydrocephalus ex vacuo, or ventricular dilation resulting from loss of cortex. In the neuropil of the cerebral cortex there is fragmentation of neurites (neuronal processes) within gray matter producing the characteristic "senile plaques“. These are degenerative presynaptic endings. The plaques may also contain a few astrocytes, and microglia. Bielschowsky silver stain. The thioflavin stain viewed with fluorescence microscopy highlights the neuritic plaques of Alzheimer's disease with amyloid deposition which fluoresces bright green, as shown here. neurofibrillary tangle Older plaques contain a central amyloid core, seen here with Congo red stain. A small vessel demonstrates amyloid angiopathy as well. • The pathologic changes of Alzheimer disease occur in nearly all Down syndrome (trisomy 21) patients by the age of 45. • Amyloid precursor protein (APP) – Abnormal APP processing leads to deposits of insoluble β-pleated amyloid protein resulting in fibrillar aggregates of beta-amyloid that is toxic to neurons. • Normal aging – Diffuse plaques and neuritic plaques: uncommon (20% of those studied) in 6th decade, common (90%) in centenarians – Neurofibrillary degeneration: uncommon in cortex in normal aging. Alzheimer disease: Summary • Most common cause of dementia in the elderly • Degenerative cell loss leads to gross atrophy of the entire brain with “hydrocephalus ex vacuo” • Neuritic plaques, neurofibrillary tangles, cell loss and gliosis, granulovacuolar degeneration, amyloid angiopathy, Hirano bodies. Pick disease • Onset 45-65 years, rare after 75 • 80% are sporadic / 20% familial • 5-10 year duration • Frontal lobe symptoms (personality changes, ‘haphazard’ behavior, lack of planning, antisocial, obsessive compulsive, language deficits, anomia and echolalia), • Anterior temporal lobe symptoms (aphasia, semantic memory loss, Kluver-Bucy syndrome) and pathology – memory relatively retained. • Gross appearance: severe localized atrophy of frontal lobes and anterior temporal lobes “knife-edge” atrophy • Microscopic appearance: Pick cells (ballooned neurons) and Pick bodies with cell loss and gliosis • Pick cells are seen in 1/2 of cases with typical gross appearance and Pick bodies only 20% Pick's disease with the gross appearance of lobar atrophy is seen here involving the frontal lobe. Note the "knife like" gyri. Pick's disease is demonstrated grossly in this coronal section in which there is marked atrophy with ex vacuo ventricular dilation. Multi-infarct dementia (MID) • Multi-infarct dementia (MID) can cause a dementia similar to Alzheimer's disease (AD). • However, no pathologic findings are present characteristic of AD. • Instead, there are multiple ischemic lesions in the cerebral cortex that cumulatively result in loss of enough neurons to produce dementia. • Most patients with MID have an abrupt onset of cognitive symptoms along with an incremental loss of mental function. • Focal neurologic deficits can be present, depending upon the size and location of the infarcts. • In some cases, though, there is gradual loss of mental function. • Pathologically, – marked cerebral arterial atherosclerosis and/or – thromboembolic disease can account for the appearance of many infarcts, typically small and scattered. Grossly, this composite view of the brain demonstrates multiple remote cystic infarcts in various locations. This process took several years. Degenerative Diseases of Basal Ganglia and Brainstem Movement disorders • Akinetic-rigid – Parkinson disease (PD) Postencephalitic parkinsonism (PEP) – Multiple system atrophy (MSA) Parkinson disease • Idiopathic Parkinson disease (vs. parkinsonism or parkinsonian syndrome) • Tremor (rest) • Rigidity (cogwheel rigidity) • Bradykinesia (mask-like facies, loss of arm-swing) • Festinating gait (loss of righting reflexes) • Gross: loss of pigment in the substantia nigra • Microscopic : Lewy bodies with pigmented neuronal cell loss and gliosis – cortical Lewy bodies present in 80% or more of PD cases • Most cases are sporadic. • Mechanism: Loss of dopaminergic input from substantia nigra to striatum • This syndrome covers several diseases of different etiologies which affect primarily the pigmented neuronal groups including the – – – – substantia nigra, locus ceruleus, dorsal motor nucleus of cranial nerve X, the substantia innominata. • The pigmented neurons are slowly lost as the disease progresses and melanin pigment can be seen within the background neuropil or within macrophages. • Astrocytosis occurs secondary to neuronal loss. • Some patients with Parkinsonian symptoms also have dementia, and in these patients there are Lewy bodies in the cerebral cortex, as well as the substantia nigra. Pathology • Lewy bodies in association with Parkinson's disease are found within the cytoplasm of pigmented neurons. • For a diagnosis of DLB, the Lewy bodies must be found in the neocortex. – These are homogeneous pink bodies on H&E stains with a surrounding halo. – Immunohistochemical staining with antibody to alpha-synuclein is positive in these Lewy bodies. A normally pigmented substantia nigra The patient with Parkinson's disease has decreased neurons and pigment as seen microscopically at the right Other parkinsonian syndromes • Differential diagnosis of parkinsonism – Progressive supranuclear palsy (PSP) – Corticobasal degeneration (CBD) – Multiple system atrophy (MSA) • striatonigral degeneration • olivopontocerebellar atrophy • Shy-Drager syndrome – Dementia pugilistica – Postencephalitic parkinsonism (PEP) Parkinson disease summary • Akinetic movement disorder • Loss of dopaminergic input from substantia nigra to striatum • Pigmented (neuromelanin containing) nerve cell loss with gliosis in the substantia nigra pars compacta and locus ceruleus (pigment loss is grossly apparent) • Lewy body formation in brainstem and cortex. Parkinson disease and dementia • Relatively common disorder in the middleaged and elderly; • The major pathologic changes are seen in the pigmented neurons of the substantia nigra in the midbrain; – neurons in this area contain intracytoplasmic inclusions called Lewy bodies. • 40% of Parkinson Disease patients develop dementia; • demented PD patients have more Alzheimer disease pathology than nondemented PD patients – some demented PD patients have only Lewy body pathology Dementia with Lewy Body Diseases (DLB) Dementia with Lewy bodies is a clinicopathological syndrome that may account for up to 20% of all cases of dementia in older patients, typically in their seventh and eighth decades. DLB There are three major syndromes associated with the appearance of Lewy bodies. These are: • (1) Parkinson disease, • (2) autonomic nervous system failure, • (3) dementia. • The clinical presentation of Lewy body disease varies according to the site of Lewy body formation and associated neuronal loss. • In Parkinson disease, the Lewy bodies are found in the substantia nigra of the midbrain, coupled with the loss of pigmented neurons. • In persons with the dementia of diffuse Lewy body disease, there are Lewy bodies in the neocortex. • Some persons have the Lewy bodies in both locations. • The basal ganglia and diencephalon may also be involved in some cases. Lewy bodies • spherical, intraneuronal, cytoplasmic, eosinophilic inclusions. H&E stain demonstrates a rounded pink cytoplasmic Lewy body in a neuron of the cerebral cortex from a patient with diffuse Lewy body disease, which can be a cause for dementia. Lewy bodies can also be seen in substantia nigra with Parkinson's disease. An immunoperoxidase stain for ubiquitin, seen at the right, helps demonstrate the Lewy bodies more readily by the brown reaction product within them. Multiple system atrophy (MSA) • Prevalence: 8% of parkinsonian cases in PD brain banks • Microscopic finding: oligodendroglial cytoplasmic inclusions • Clinical signs and symptoms – Parkinsonism (striatonigral degeneration) – Autonomic failure (Shy-Drager syndrome) – Cerebellar findings (olivopontocerebellar degeneration) • Striatonigral degeneration – parkinsonism with orthostatic hypotension and cerebellar ataxia – cell loss and gliosis in putamen • Olivopontocerebellar atrophy (sporadic) – neuronal loss and gliosis • Shy-Drager syndromesympathetic ganglia Movement disorders • AkineticPD, other diseases with parkinsonian features • Hyperkinetic – Huntington chorea – Other • Myoclonus • Ballismus • Dystonia • Tic disorders Huntington’s disease • Choreiform movements, neuropsychiatric disturbance and slowly progressive dementia – seizures or rigidity may occur initially • Gross: atrophic, flattened caudate • Microscopic: cell loss and gliosis in caudate nucleus (dorsal putamen, globus pallidus and nucleus accumbens affected later) Huntington's disease is shown grossly in this coronal section of the brain. It demonstrates atrophy of the caudate with resultant increase in size of lateral ventricles. Microscopically, the caudate nucleus in Huntington's disease demonstrates loss of neurons along with gliosis. • Autosomal dominant genetic disorder, (sporadic mutations not described) • Characterized by chorieform movements, cognitive deterioration, emotional disturbances • Huntingtin gene (HD gene), chromosome 4, contains an unstable trinucleotide repeat (CAG= 37-86 vs 19 in normals, codes for glutamic acid) • Complete dominance-- heterozygotes as severely affected as homozygotes • Children inheriting the gene from their father have either juvenile onset disease or onset approximately 3 years early (genomic imprinting) • Onset is typically in 4-5 decade Huntington Disease: Summary • Hyperkinetic movement disorder • Characterized by chorieform movements, cognitive deterioration, emotional disturbances • Caudate atrophy • Deep gray nuclei cell loss/gliosis • Autosomal dominant • Trinucleotide repeat disease * * * *N. Caudatus *Corpus callosum *Thalamus Basal ganglia • Typical disorders of the basal ganglia, (dyskinesias) include: • Huntington chorea--striatum, • ballismus--subthalamic nucleus and • PD--substantia nigra • . • The functional definition of the basal ganglia includes: – the corpus striatum (lentiform and caudate nuclei), – subthalamic nucleus. – substantia nigra. SPINOCEREBELLAR DEGENERATIONS • Spinocerebellar Ataxias • Friedreich Ataxia • Ataxia-Telangiectasia Spinocerebellar Ataxias • This is a group of genetically distinct diseases characterized by signs and symptoms referable to the cerebellum (progressive ataxia), brainstem, spinal cord, and peripheral nerves, as well as other brain regions in different subtypes. • Pathologically, they are characterized by neuronal loss from the affected areas with secondary degeneration of white matter tracts. Friedreich Ataxia • This is an autosomal-recessive progressive , beginning in the first decade of life with gait ataxia, followed by hand clumsiness and dysarthria. • Deep tendon reflexes are depressed or absent, but an extensor plantar reflex is typically present. • Joint position and vibratory sense are impaired, and there is sometimes loss of pain and temperature sensation and light touch. • Most patients develop pes cavus and kyphoscoliosis. • There is a high incidence of cardiac disease with arrhythmias and congestive heart failure. • Concomitant diabetes is found in about 10% of patients. • Most patients become wheelchair-bound within about 5 years of onset; the cause of death is intercurrent pulmonary infections and cardiac disease. Ataxia-Telangiectasia • Ataxia-telangiectasia is an autosomal-recessive disorder • characterized by an ataxic-dyskinetic syndrome • beginning in early childhood, • caused by neuronal degeneration predominantly in the cerebellum, • the subsequent development of telangiectasias in the conjunctiva and skin, and immunodeficiency. DEGENERATIVE DISEASES AFFECTING MOTOR NEURONS • Lower motor neurons in the anterior horns of the spinal cord • Lower motor neurons in certain cranial nerve motor nuclei (V, VII, IX, XII) but not those that control eye movements (III, IV, VI) • Upper motor neurons (Betz cells) in the motor cortex DEGENERATIVE DISEASES AFFECTING MOTOR NEURONS • Amyotrophic Lateral Sclerosis (Motor Neuron Disease) • Bulbospinal Atrophy (Kennedy Syndrome) • Spinal Muscular Atrophy • Genetic Metabolic Diseases Amyotrophic Lateral Sclerosis ALS • ALS – (also known as Lou Gehrig's disease after the famous Yankee first baseman who had this disease) • results from loss of motor neurons which is most striking in the anterior horn cells of spinal cord but may involve cranial motor nuclei and Betz cells(large pyramidal cells in the motor cortex of the precentral gyrus). • The loss of anterior horn cells leads to muscle atrophy. • Males are affected more commonly than females. • The patients present in middle age with weakness of the extremities and may go on to develop bulbar signs and symptoms. • Astrocytosis is seen in response to the loss of motor neurons. • Because of the loss of upper motor neurons, there is lateral column degeneration with gliosis, the so-called "sclerosis" of the lateral columns of spinal cord. • The course is usually 2 to 6 years after diagnosis, – but patients presenting with bulbar signs and symptoms have a shorter life span because of swallowing difficulties and aspiration. • The etiology is unknown. • Most cases occur sporadically, – but 1 to 10% of cases may have an autosomal dominant inheritance pattern. There is lateral column degeneration of the spinal cord microscopically in this case of ALS, as seen with Luxol fast blue stain. Bulbospinal Atrophy (Kennedy Syndrome) • This X-linked adult-onset disease is characterized by distal limb amyotrophy and bulbar signs such as atrophy and fasciculations of the tongue and dysphagia. • Affected individuals manifest androgen insensitivity with gynecomastia, testicular atrophy, and oligospermia. Genetic Metabolic Diseases • Neuronal storage diseases are mostly autosomalrecessive diseases caused by a deficiency of a specific enzyme involved in the catabolism of sphingolipids, mucopolysaccharides, or mucolipids. • Leukodystrophies show a selective involvement of myelin (either abnormal synthesis or turnover), and generally exhibit no neuronal storage defects. • Mitochondrial encephalomyopathies are a group of disorders of oxidative phosphorylation, usually resulting from mutations in the mitochondrial genome LEUKODYSTROPHIES • Krabbe Disease This disease is an autosomal-recessive leukodystrophy resulting from a deficiency of galactocerebroside β-galactosidase (galactosylceramidase), the enzyme required for the catabolism of galactocerebroside to ceramide and galactose LEUKODYSTROPHIES • Metachromatic Leukodystrophy • This disorder is transmitted in an autosomal-recessive pattern and results from a deficiency of the lysosomal enzyme arylsulfatase LEUKODYSTROPHIES • Adrenoleukodystrophy • This disorder, which has several clinically and genetically distinct forms, is a progressive disease with symptoms referable to myelin loss from the CNS and peripheral nerves as well as adrenal insufficiency. • In general, forms with earlier onset have a more rapid course. • The X-linked form usually presents in the early school years with neurologic symptoms and adrenal insufficiency and is rapidly progressive and fatal • Dementia – – – – Summary Prion disease Alzheimer’s disease Dementia with Lewy bodies Pick’s lobar atrophy • Movement disorders – Akinetic rigid--Parkinson disease and parkinsonian syndromes – Hyperkinetic--Huntington disease Prion disease • Spongiform encephalopathy – Kuru (New Guinea) – Creutzfeldt-Jakob (sporadic, iatrogenic) – Gerstmann-Sträussler-Scheinker syndrome • familial, ataxia prominent – FFI (fatal, familial insomnia) – Animals: BSE ( bovine spongiform encephalopathy), scrapie TRANSMISSIBLE AGENT (PRION) DISEASES SPONGIFORM ENCEPHALOPATHY CREUTZFELDT-JACOB DISEASE • 50-70 years old, rapidly evolving dementia, – myoclonus – characteristic EEG pattern of repetitive sharp waves. Early symptoms include personality changes, impaired judgement, gait abnormalities, vertigo. • In some patients cerebellar and visual abnormalities predominate • Majority die within 3-6 months. Prion disease pathogenesis • Transmissible but not “infectious” • Prion protein, Prusiner--1997 Nobel Prize, (not a “slow virus”, just a protein). • PrPC produced normally in most cells amino acid sequence abnormal protein. • Abnormal PrPC deposits in specific locations causing specific clinical features (thalamus in FFI-- fatal, familial insomnia), • Localization correlates with clinical findings: – cerebellar findings with cerebellar “Kuru plaques” in Gerstmann-Sträussler-Scheinker syndrome. • Gross appearance – may be normal due to short duration of disease • Microscopic appearance – Vacuolation of neuropil*, within nerve cell bodies and neuronal processes: • Many round to oval vacuoles varying in size from one to 50 microns in size in the neuropil of cortical gray matter. • These vacuoles may be single or multiloculated. • The vacuoles may coalesce to microcysts. *Neuropil: a dense network of neurons and glia in the central nervous system; the brain tissue that lies between axons. – Cell loss and gliosis may be prominent. There are numerous gray matter vacuoles, along with gliosis and neuronal loss, in a patient with advanced CJD. Prion disease: Summary • • • • A rapidly progressive dementing disease EEG characteristic Gross brain often not atrophic Spongiform changes on microscopic examination (gliosis may be extensive) • Caused by a abnormal protein (PrPC) not a virus.