Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Designer baby wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Microevolution wikipedia , lookup
Maximum parsimony (phylogenetics) wikipedia , lookup
Genome evolution wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Metagenomics wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Sequence alignment wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Gene expression programming wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
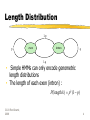
Comp. Genomics Recitation 9 11/3/06 Gene finding using HMMs & Conservation Outline • Gene finding using HMMs • Adding trees to HMMs • phyloHMM • N-SCAN • BLAST+ Gene Finding • SGP2 • Examples Markov Sequence Models • Key: distinguish coding/non-coding statistics • Popular models: • 6-mers (5th order Markov Model) • Homogeneous/non-homogeneous (reading frame specific) • Not sensitive enough for eukaryote genes: exons too short, poor detection of splice junctions 3 Length Distribution 1-p p exon intron q 1-q • Simple HMMs can only encode genometric length distributions • The length of each exon (intron) : P(length k ) p k (1 p) CG © Ron Shamir, 2008 4 Exon Length Distribution • The length distribution of introns is ≈ geometric • For exons, it isn’t: also affected by splicing itself: • Too short (under 50bps): the spliceosomes have no room • Too long (over 300bps): ends have problems finding each other. • But as usual there are exceptions. • A different model for exons is needed • A different model is needed for exons. CG © Ron Shamir, 2008 5 Generalized HMM (Burge & Karlin, J. Mol. Bio. 97 268 78-94) • Instead of a single char, each state omits a sequence with some length distribution CG © Ron Shamir, 2008 6 Generalized HMM (Burge & Karlin, J. Mol. Bio. 97 268 78-94) • Overview: • Hidden Markov states q1,…qn • State qi has output length distribution fi • Output of each state can have a separate probabilistic model (weight matrix model, HMM…) • Initial state probability distribution • State transition probabilities Tij CG © Ron Shamir, 2008 7 GenScan Model CG © Ron Shamir, 2008 Burge & Karlin JMB 97 8 GenScan model • states = functional units on a gene • The allowed transitions ensure the order is biologically consistent. • As an intron may cut a codon, one must keep track of the reading frame, hence the three I phases: • phase I0: between codons • phase I1:: introns that start after 1st base • phase I2 : introns that start after 2nd base CG © Ron Shamir, 2008 9 Phylogenetic HMMs • Due to Siepel and Haussler • A simple gene-finding HMM looks at a single Markov process: • Along the sequence: each position is dependent on the previous position • If we incorporate sequences from multiple organisms, we can look at another process: • Along the tree: each position is dependent on its ancestor Phylogenetic HMMs • A simple HMM can be thought of as a machine that generates a sequence • Every state omits a single character • Multinomial distribution at every state • A phyloHMM generates an MSA • Every state omits a single MSA column • Phylogenetic model at every state Phylogenetic HMMs Phylogenetic models in phyloHMM • Defines a stochastic process of substitution • Every position is independent • The following process occurs: • A character is assigned to the root • The character substitution occur based of some substitution matrix and based on the branch lengths • The characters at the leaves of the tree correspond to the MSA column Phylogenetic models in phyloHMM • Different models for different states: • Different substitution rates • E.g., in exons, we’ll see less substitutions • Different patterns of substitutions • E.g., third position bias in coding sequences • Different tree topologies • E.g., following recombination Formally • S – set of states • Ψ – phylogenetic models (instead of E in a standard HMM) • A – state transitions • b – initial probabilities Formally • Q – substitution rate matrix (e.g., derived from PAM) • Π – background frequencies • τ – the phylogenetic tree • β – branch lengths Formally • - Probability of a column Xi being omitted by the model ψi • Can be computed efficiently by Felsenstein’s “pruning algorithm” (recitation 6) • Joint probability of a path in the HMM and and alignment X • Viterbi, forward-backward etc. – as usual Simple phylo-gene-finder Non-coding 3rd position • If the parameters are known – Viterbi can be used to find the most probably path – segmentation into coding regions Phylo-gene-finder is a good idea • Use of phylogeny is important: • Imposes structure on the substitutions • Weights different pairs differently based on the evolutionary distance N-SCAN • Another phylogeny-HMM-gene-finder • A GHHM that emits MSA columns • Annotates one sequence at a time: the target sequence • Distinguishes between a target sequence – T and other informative sequences (Is) that may contain gaps • States correspond to sequence types in the target sequence N-SCAN • Bayesian network instead of a simple evolutionary model • Accounts for: • 5’ UTRs • Conserved non-coding • Highly conserved • No “coding” features SGP-2 • Drawback of the described approaches: require meaningful alignment • Impossible if one of the genomes is not yet finished • An alignment is not necessary “correct” SGP-2 • A framework working on two genomes • Idea: • Use BLAST to identify which positions are more/less conserved • Feed the BLAST scores into the gene-finding HMM • The BLAST results serve to modify the scores of the exons. SGP-2 BACH1 OLIG2 PPM1A Summary • Different approaches for gene finding • Adding phylogeny generally helps • But • What about genes/exons which are specific to humans • Ape genomes are not (almost) available and too similar • Phylogenetic help almost essential in more difficult problems • Motif finding (promoter analysis) • Ultraconserved regions with no evident function