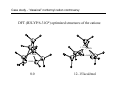
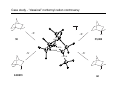
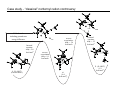
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Metallic bonding wikipedia , lookup
Stoichiometry wikipedia , lookup
Molecular Hamiltonian wikipedia , lookup
Green chemistry wikipedia , lookup
Solvent models wikipedia , lookup
Condensed matter physics wikipedia , lookup
Nuclear chemistry wikipedia , lookup
Metastable inner-shell molecular state wikipedia , lookup
Hartree–Fock method wikipedia , lookup
Electronegativity wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Photoredox catalysis wikipedia , lookup
History of molecular theory wikipedia , lookup
Marcus theory wikipedia , lookup
Inorganic chemistry wikipedia , lookup
Click chemistry wikipedia , lookup
History of chemistry wikipedia , lookup
Nanochemistry wikipedia , lookup
Chemical bond wikipedia , lookup
Atomic orbital wikipedia , lookup
Atomic theory wikipedia , lookup
Jahn–Teller effect wikipedia , lookup
Transition state theory wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
Photosynthetic reaction centre wikipedia , lookup
Hypervalent molecule wikipedia , lookup
Molecular orbital wikipedia , lookup
Resonance (chemistry) wikipedia , lookup
Analytical chemistry wikipedia , lookup
Bent's rule wikipedia , lookup
Coupled cluster wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Electron configuration wikipedia , lookup
Molecular orbital diagram wikipedia , lookup
Introduction to Computational Chemistry WISOR XI Bressanone, Italy, January 6-13, 2002 Peter R. Schreiner Hans-Ullrich Siehl Computational Center for Molecular Structure and Design University of Georgia, Department of Chemistry Athens, GA 30602-2556 [email protected] Abteilung Organische Chemie I Universitaet Ulm D-89069 Ulm, Germany [email protected] What is Computational Chemistry? ”Computational chemistry is to model all aspects of chemistry by calculation rather than experiment." Paul v. R. Schleyer (1987) "Computational Chemistry has existed for half a century, growing from the province of a small nucleus of theoretical work to a large, significant component of scientific research. By virtue of the great flexibility and power of electronic computers, basic principles of classical and quantum mechanics are now implemented in a form which can handle the many-body problems associated with the structure and behavior of complex molecular systems." John A. Pople (November 1997) (Nobel prize for chemistry 1998, together with Walter Kohn ) Why do chemistry (an “experimental” science?) on computers? • Improved chemical (physical) understanding • Faster turnaround of ideas (not always) • Reduced cost (and waste!) • Safety • Better accuracy (for very small systems) Computational chemistry is far from black box! Care must be taken in choice of method. Elements of Computational Chemistry • Decipher the “language” (many different approaches with even more acronyms – learn) • Technical aspects – how to transfer an idea (molecular structure or property, chemical reaction, etc.) into the program and the computer (in a reasonable time frame) • Quality assessment: most important, most difficult! Required: • Understand the basis of the underlying theoretical concepts of a particular approach to solving the problem • Understand the chemistry (above all!) • Constantly compare experiment and theory (be critical of both) Conceptual approach to computational chemistry Prediction Interpretation Validation "...give us insight, not numbers." C. A. Coulson (Proc. Robert A. Welch Foundation Conf. XVI, S. 117) Chemistry is knowing the energy as a function of the nuclear coordinates. supplied • graphically • "by hand" molecule coordinates • cartesian • internal different types for different purposes human input: choice! difficult program molecular properties interpre • structures • energies • molecular orbitals • AMBER, CHARMM, GROMOS, Sybyl... • IR, NMR, UV... • AMPAC, MOPAC, VAMP... • Gaussian, Gamess, MOLPRO, Jaguar, PSI... many different ones: (—> interpretation) (—> different strengths & purposes) (—> different models) Describing properties is knowing how the energy changes upon adding a perturbation. Computational chemistry methods • Molecular mechanics, force fields easy to comprehend quickly programmed extremely fast no electrons: limited interpretabilty • ab initio methods full quantum method only expt. fundamental constants very high accuracy complete (all interactions are included) very time consuming (“expensive”) systematic improvement possible • Semiempirical methods quantum method valence electrons only fast limited accuracy • Density functional theory quantum method in principle “exact” faster than trad. ab initio variable accuracy no systematic improvement Force fields Simple harmonic oscillator model —> close relationship to potential energy surface Typical accuracy (MM2 force field of Allinger et al.): — — — — — — 0.01 Å bond lengths 1˚ bond angles few degrees for torsion angles conformational energies: accurate to 1 kcal/mol (at best). vibrational frequencies: 20 – 30 wavenumbers (at best) configurational sampling (in MD): few kcal/mol Semiempirical methods • treat valence electrons only (core potential) • minimal basis set (Slater (not Gaussian!) type orbitals, STO) • neglect all two-electron integrals involving two-center charge distributions, i.e., all three-center and four-center two-electron integrals. Replace by parameters to mimick experimental results (geometries and heats of formation). • solve secular equations just like in self-consistent field (HF) theory • typical methods: MNDO, MINDO, AM1, PM3 (order of development) Caveat: Semiempirical methods describe ground-states only and are geared towards computing the heats of formation! Semiempirical methods • Faster than ab initio or DFT: x 1000 (N2 scaling) for energies • N3 for gradient • Harmonic force fields calculated from numerical finite differences of gradients • Treat up to 10000 heavy (= non-hydrogen) atoms! Accuracy and limitations (organic molecules): • mean absolute deviations: 6.3 kcal/mol for heats of formation • 0.014 Å for bond lengths • 2.8° for bond angles • 0.48 eV for first ionization potentials Some qualitative deficiencies: • Sterically crowded molecules are too unstable (e.g., neopentane vs. n-pentane). • Four-membered rings are too stable (e.g. cyclobutane, cubane). • Non-classical structures are normally too unstable. • Rotational barriers are often underestimated (e.g. ethane)! • Hydrogen bonds are much too weak and too long (e.g., water dimer)! • Activation barriers are normally somewhat too high. • Pericyclic reactions: biradicaloid mechanisms are artificially favored. Hartee Fock (self-consistent field, SCF) theory: key steps Schrödinger equation: Space & time separation: non-rel. Hamilton operator: Born-Oppenheimer approximation: Hψ = Eψ ψ(r, t) = ψ(r) ψ(t) —> Hψ (r) = Eψ (r) H = T + V = –(h2∇2/8pm) + V ψ (r, R) ≈ ψ (r) ψ (R) +∞ 2 Normalization: |cΨ| dτ = 1 -∞ Antisymmetrization: Molecular Orbitals: ψ (r1,...ri,rj,...rn) = – ψ (r1,...rj,ri,...rn) ψ (r) = φ1 (r1) φ2 (r2) ...φn(rn) Ν LCAO: φi = ∑ cµiχµ µ=1 Basis functions: gs(α, r) = Cxnymzlexp(-αr2) Variation principle: E (ψi) > E (ψ) with ψi ≠ ψ HF/SCF quality assessment: general aspects • Bond lengths and angles of "normal" organic molecules quite accurate (within 2%) • Conformational energies accurate to 1–2 kcal/mol. • Vibrational frequencies for most covalent bonds systematically too high by 10–12% • Zero point vibrational energies: ~1-2 kcal/mol • Isodesmic reaction energies accurate to 2–5 kcal/mol. • Entropies accurate to 0.5 cal/K mol. • Protonation/Deprotonation energies ~10 kcal/mol (gas phase) • Atomization and homolytic bond-breaking reactions: (25–40 kcal/mol) • Reaction barriers may (!) have large errors. HF/SCF quality assessment: geometries and rotational barriers Rotational barriers Bond lengths and angles Molecule H3B-NH3 STO-3G 3-21G 6-31G** Expt. 2.1 2.9 1.9 2.7 1.9 3.0 3.1 2.9 2.8 2.0 2.0 1.5 2.4 1.4 2.0 1.1 1.3 1.9 1.1 1.7 1.4 2.0 1.7 2.0 HO-OH cis 1.5 9.1 1.1 11.7 1.4 9.2 1.3 7.0 HS-SH cis 6.1 5.7 8.5 6.8 Basis ∆r (Å) Angle (°) STO-3G 3-21G 0.054 0.016 2.3 3.8 3-21G(*) 0.017 3.3 6-31G* 0.014 1.5 H3C-OH H3C-SiH3 6-31G** 6-311G** 0.014 0.013 1.8 1.0 H3C-PH2 H3C-SH H3C-CH3 H3C-NH2 HF/SCF quality assessment: isomerization energies Formula Reaction HF/6-31G* Experiment HCN Hydrogen cyanide —> Hydrogen isocyanide 12.4 14.5 CH2O Formaldehyde —> Hydroxymethylene 52.6 4.9 CH3NO Formamide —> Nitrosomethane 65.3 62.4 C2H3N Acetonitrile —> Methyl isocyanide 20.8 20.9 C2H4O Acetaldehyde —> Oxacyclopropane 33.4 26.2 C3H6 Propene —> Cyclopropane 8.2 6.9 Classical failures of HF theory: •FOOF: R.D. Amos, C.W. Murray, and N.C. Handy, Chem. Phys. Lett. 202, 489 (1993). •Cr-Cr: G.E. Scuseria, J. Chem. Phys. 97, 7528 (1992). •NO3: P.S. Monks, et al., J. Phys. Chem. 98, 10017 (1993). Beyond HF/SCF: electron-correlation methods HF/SCF: electrons interact with each other only via the average electron density. Reality: instantaneous repulsion = correlation energy. Most common methods to include electron correlation: • Perturbation Theories (MPn) • Configuration Interaction (CI) • Coupled Cluster (CC) • Density Functional Theory (DFT) Correlation methods: Møller-Plesset perturbation theory —> non-iterative corrections added to the Hartree-Fock energy H = Ho + λH1 —> perturbation is small, H1 can be expressed in a series in terms of the parameter λ: ψ = ψ(0) + λ1ψ(1) + λ2ψ(2) + λ2ψ(2) + ... E = E(0) + λ1E(1) + λ2E(2) + λ3E(3) + ... Problems: • overcorrection possible • non-convergent series Correlation methods: configuration interaction —> wavefunction expressed as a linear combination of all possible HF-determinants: n ψ = b0 ψ0 + ∑ bsψs s>0 HF S S D D Problems: • very time-consuming • not size-extensive • slow convergence • strongly basis set-dependent T Q Correlation methods: coupled-cluster theory —> tries to include all corrections (S, D, T, ...) to infinite order —> operator T performs changes of the electron distribution, as expressed through the molecular orbitals φi to compute the correlation energy: ψ = eTφo H eTΨ0 = E eTΨ0 —> single, double…etc substitutions (CCD, CCSD, CCSD(T)…) —> CCSD(T) is most common and probably the best choice as the triple contributions are included perturbatively through an additional term arising from 5th order MP theory. —> QCISD and QCISD(T) are similar to CCSD and CCSD(T), respectively, but some of the terms have been omitted. As there is virtually no computation advantage of the CI methods in this context, QCI should not be used! Correlation improves dissociation energies dramatically Reaction H2 -> H•+H• HF/6-31G** MP2/6-31G** MP3/6-31G** MP4/6-31G** Expt. 85 45 52 101 48 52 105 49 49 106 58 47 109 32 50,56 FH -> F•+H• 3CH2 -> CH•+H• 1CH2 -> CH•+H• 93 101 131 109 127 108 128 107 141 105,106 70 89 90 91 98 OH2 -> OH•+H• BH3 -> BH2•+H• 86 90 83 119 106 110 115 108 108 116 109 109 126 57-107 116 87 109 110 110 113 LiH -> Li•+H• BeH -> Be•+H• NH3 -> NH2•+H• CH4 -> CH3•+H• Correlation methods: scaling Method Ave. Error (kcal/mol) vs. FCI approx. time factor HF DFT MP2 5-30 2–10 17.4 ON2-3 ON3 ON4 MP3 MP4 MP5 14 4 3.2 ON5 CISD CCSD CCSD(T) 13.8 4.4 CCSDT CCSDTQ 0.5 0.0 ON6 O2N7 O2N>7 O2N>>7 Correlation methods: systematic quality improvement Basis set minimum HF DZ DZP TZP QTZ ... infinite HF limit Method CID CISD CISDT CISDTQ FCI "exact" Density functional theory (DFT) Hohenberg-Kohn theorem: energy and all other electronic properties of the ground state are uniquely determined by its charge density ρ(r): EV [ρ ]= FHK + ∫ ρ(r )V (r )dr • What is the form of FHK (functional)? • How can the exact charge density ρ(r) be determined? One particle eigenvalue: effective one electron Hamilton (UHF!), HKS, which incorporates both, FHK[ρ] and V: 1 ZA H φ (r1 ) = − ∆ + ∑ + 2 R − r A A 1 γ K S iσ ∫ ρ γ (r2 ) + VXC (r ) φ iσ (r1 ) = ε iσ φ iσ (r1 ) r1 − r2 Density functional theory (DFT) • Self consistent solutions for φiσ resemble those of HF theory, but DFT orbitals have no physical significance other than constituting the charge density (still being discussed). • DFT wavefunction is not a Slater determinant of spin orbitals! In a strict sense there is no N-electron wave function available in DFT! • KS equations give exact solution of N-electron problem, provided the correct functional dependence of exchange correlation EXC energy with respect to the charge density ρ(r) is available. Density functional theory performance F• + H2 —> FH + H• Re barrier Cr2 properties De vib. HF 14.2 1.465 –19.4 1151 CCSD CCSD(T) 3.3 2.3 1.560 1.621 –2.9 0.5 880 705 DFT 3.6 1.590 1.5 597 Expt. 2.0 1.679 1.4 470 Wave function interpretation Why can the wave function not be interpreted directly? ψ = φ 1φ 2... φ n —> with φ i = ∑ cir χr r and χr = ∑ arn ξn n results in a list of numbers without obvious chemical meaning. The electron density ρ does have physical meaning: N/2 ρ(r) = ψ(r) 2 with ρ(r) = 2∑ φ(r) i N/2 2 and dr ρ(r) = 2∑ Problem: ρ is four-dimensional (number of electrons plus x,y,z coordinates). Presentation and interpretation is difficult. "Atomic" information is desireable: Charges, bonds, dipoles, etc. i dr φ(r) 2= N Wave function interpretation Filter (translate into chemical lingo) out partial information Solution: a) Density methods (do something with the density so it can be interpreted): • • • b) Bader’s “Atom in Molecules” (density gradients) Electrostatic potentials (molecular polarization) Density surface (spatial extension, shape analysis) Molecular orbital methods (do something with the orbitals so they can be interpreted): • • • Mulliken population analysis Löwdin population analysis Natural population analysis, natural bond orbital analysis Mulliken population analysis Distribution of electrons based on occupations of atomic orbitals (AOs), φi. Principle: N= MO's AO's ∑ ∑r i N(i) AO's MO's cir2 +2 Atomic part ∑ i N(i) ∑ r>s cir cis Srs overlap contributions But: Atomic population n(k) does not include electrons from the overlap population N(k,l)! Mulliken: Divide overlap population exactly in the middle, regardless of type, EN, etc.: MO's N(k) = ∑ i N(i) ∑ cir k cir k + ∑ cis lSr ksl rk sl≠k Mulliken, R. S. J. Chem. Phys. 1955, 23, 1833, 1841, and 2338. with qk = Zk – N(k) Mulliken population analysis Advantages: • easy to program • fast • commonplace (easy for comparisons) Drawbacks: • differences of atom types are not properly accounted for • strong basis set dependence • N(k) may be larger than 2: violation of Pauli-principle Remedy: Lödwin analysis • Symmetric transformation of Mulliken-analysis in orthogonal coordinate system so that 0 < N(k) < 2 Löwdin, P.-O. Adv. Quantum Chem. 1970, 5, 185. Natural population analysis / natural bond orbital methods (Weinhold et al.) Principle: Uses orbitals which diagonalize the one-particle density matrix (natural orbitals, NO). Diagonalisation of the two-center subdeterminants gives the "natural bond orbitals." Advantages: • very detailed analysis possible • little basis set dependent • atomic properties fully included Drawbacks: • more time consuming than simpler methods (unimportant, though) • sometimes numerically unstable • highly delocalized are troublesome Reed, A. E.; Weinstock, R. B.; Weinhold J. Chem. Phys. 1985, 83, 735. Examples: LiH charges with Mulliken and NPA analyses Level Li (LiH) Li (LiF) HF/STO-3G -0.016 +0.229 HF/6-31G* +0.170 +0.666 HF/6-311+G** +0.358 +0.718 HF/STO-3G +0.425 +0.493 HF/6-31G* +0.731 +0.917 HF/6-311+G** +0.815 +0.977 Mulliken NPA Examples: Diborane (B2H6) HF/STO-3G ±0.00 (Mulliken) –0.01 (NPA) HF/3-21G ±0.02 (Mulliken) +0.08 (NPA) HF/STO-3G +0.08 (Mulliken) –0.20 (NPA) HF/3-21G –0.09 (Mulliken) –0.04 (NPA) NBO Bond order: 0.59 (HF/STO-3G) ! 0.61 (HF/6-31G*) ! HF/STO-3G –0.04 (Mulliken) –0.09 (NPA) HF/3-21G ±0.02 (Mulliken) +0.02 (NPA) Enols and enolates: electron density, potential and charges –0.27 –0.04 –0.28 enol AM1 optimized structures; HF/6-31G* densities; NBO charges –0.60 +0.19 –0.66 enolate Charges in acetone and protonated acetone (or methylated) –0.29 –0.10 +0.22 +0.32 remember: "S N 2-like" Nu – Not the case! AM1 optimized structures; HF/6-31G* NBO charges O Charges and dipoles on typical “Hammett-systems” –0.36 +0.35 –0.35 –0.35 +0.56 +0.35 –0.35 +0.23 –0.38 –0.39 +0.36 +0.23 –0.33 –0.31 µ = 2.4 D –0.32 µ = 3.4 D –0.59 +0.36 –0.55 –0.40 +0.36 +0.57 µ = 12.5 D AM1 optimized structures; HF/6-31G* NBO charges µ = 4.5 D µ = 6.0 D –0.57 +0.20 –0.44 +0.36 µ = 13.7 D Computational limitations (as of 2002…) Good methods Difficult Not yet realized available (well under development) (often attempted) Geometries Reaction rates Crystal structures (prediction) Energies (small Circular dichroism (optical, Melting points systems) magnetic, vibrational) IR Spin-orbit couplings Protein folding NMR Full relativistics Full reaction dynamics ESR Excited states (vertical) Molecular dynamics MW Solvent effects Excited states (adiabatic) Multipole moments Density matrix methods/geminals Solvent dynamics PE-spectra Linear scaling Systematic improvement of DFT Local correlation methods r12-methods Accurate enzyme-substrate interactions Case study - “classical” norbornyl cation controversy ≠ δ− X δ+ X H H exo nonclassical + ROS ≠ +X H X endo OS + - H H X classical exo/endo (X = OBrosylate) rate ratio= 350 (AcOH) i.e. energy differences in transition states 2-5 kcal/mol H Case study - “classical” norbornyl cation controversy DFT (B3LYP/6-31G*) optimized structures of the cations: C C C 1.711ÅC 2.087Å C C 1.892Å C 1.414Å C C 1.395Å 0.0 C C C C C 12–15 kcal/mol Case study - “classical” norbornyl cation controversy X X Me -X- -X- C C 50 1.543Å C 55,000 C C 1.437Å C -X- Me 2.246Å C -X- 1.471Å C Me Me X 0.00035 X 60 Solvation — Application of MC-Methods: Example ∆∆G (kcal mol-1) 0.8 0.6 0.4 ∆∆G (kcal/mol) 0.2 0 0 0.25 0.5 0.75 1 1.25 structural parameter λ J. Am. Chem. Soc. 1995, 117, 2663 How to treat a solvolysis reaction computationally? E R+ + Xca. 200 kcal/mol R• + X• R-X reaction coordinate Problem: homolytic bond cleavage in the gas phase! How to treat a solvolysis reaction computationally? Trick: protonation leads to cation smoothly! E R+ + HX R-XH+ reaction coordinate Case study - “classical” norbornyl cation controversy C C 1.581Å C C C C C 1.449Å 2.447Å C C 1.522Å O 2.396Å C C 1.791Å mol-1 3.7 kcal including ground state energy difference forward reaction: ∆H ‡ = 2.8 kcal mol-1 C C 1.550Å C C 3.104Å 1.399Å 1.997Å C O reverse association: ∆H‡ = 5.6 kcal mol-1 TSexo C 1.514Å 2.432Å C C 1.623Å C C reverse association: ∆H‡ = 1.9 kcal mol-1 C C C C C C 1.519Å 2.527Å C 1.615Å 1.874Å C C C 1.395Å O C O O 1 (X=OH2+) (0.0 kcal mol-1) forward reaction: ∆H‡ = 5.3 kcal mol-1 C 1.560Å C C TSendo 2.356Å 6 {0.9 kcal mol-1} 2 (X=OH2+) {1.2 kcal mol-1} Case study - “classical” norbornyl cation controversy