Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Photosynthetic reaction centre wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Signal transduction wikipedia , lookup
Peptide synthesis wikipedia , lookup
Oligonucleotide synthesis wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Gene regulatory network wikipedia , lookup
Biosynthesis wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Gaseous signaling molecules wikipedia , lookup
Proteolysis wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Point mutation wikipedia , lookup
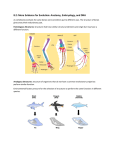
Learning Objectives: At the end of the lecture the student should be able to : 1. The process of heme synthesis and its incorporation into globin chains to form hemoglobin 2. The structure of hemoglobin from a biochemistry perspective 3. Porphyrias ,their different types ,clinical presentations and their implications 4. Various types of hemoglobin normally found in human body 5. Various types of hemoglobin associated with different diseases 6. Hemoglobin F ,its synthesis and importance of its affinity with 2,3- BPG 7. Hemoglobin A1C and its clinical implications Lecture outline: Hemoglobin synthesis • Requires co-ordinated production of heme and globin The Heme synthesis • Synthesized in a complex series of steps involving enzymes in the mitochondrion and in the cytosol of the cell • The first step in heme synthesis takes place in the mitochondrion, with the condensation of succinyl CoA and glycine by ALA synthase to form 5aminolevulic acid (ALA). • This molecule is transported to the cytosol where a series of reactions produce a ring structure called coproporphyrinogen III. • This molecule returns to the mitochondrion where an addition reaction produces protoporhyrin IX. • The enzyme ferrochelatase inserts iron into the ring structure of protoporphyrin IX to produce heme. Clinical correlation of heme synthesis • Deranged production of heme produces a variety of anemias • Iron deficiency, the world's most common cause of anemia, impairs heme synthesis thereby producing anemia. • A number of drugs and toxins directly inhibit heme production by interfering with enzymes involved in heme biosynthesis. • Lead commonly produces substantial anemia by inhibiting heme synthesis, particularly in children. The Globin synthesis • Synthesis occurs in the ribosomes present in the cytosol • Two distinct globin chains (each with its individual heme molecule) combine to form hemoglobin. • One of the chains is designated alpha. • The second chain is called "non-alpha". • With the exception of the very first weeks of embryogenesis, one of the globin chains is always alpha. • A number of variables influence the nature of the non-alpha chain in the hemoglobin molecule. • The combination of two alpha chains and two non-alpha chains produces a complete hemoglobin molecule (a total of four chains per molecule). Structure of Hemoglobin • Hemoglobin exhibits characteristics of both the tertiary and quaternary structures of proteins. • Most of the amino acids in hemoglobin form alpha helices, connected by short non-helical segments • Hydrogen bonds stabilize the helical sections inside this protein, causing attractions within the molecule, folding each polypeptide chain into a specific shape. • Hemoglobin's quaternary structure comes from its four subunits in roughly a tetrahedral arrangement. • In most humans, the hemoglobin molecule is an assembly of four globular protein subunits • Each subunit is composed of a protein chain tightly associated with a non-protein heme group • Each protein chain arranges into a set of alpha-helix structural segments connected together in a globin fold arrangement. • This is so called because this arrangement is the same folding motif used in other heme/globin proteins such as myoglobin. Adult human hemoglobin • In adult humans, the most common hemoglobin type is a tetramer (which contains 4 subunit proteins) called hemoglobin A • It consists of two α and two β subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively • This is denoted as α2β2 • Each subunit has a molecular weight of about 17,000 daltons, for a total molecular weight of the tetramer of about 68,000 daltons (64,458 g/mol) Porphyrias Group of disorders due to abnormalities in genes directing enzymes in bio-synthesis of heme. Autosomal dominant in inheritance, except: congenital erythropoietic porphyria which is autosomal recessive. Classification of Porphyrias • Acute Porphyrias • Alanine D deficient porphyria. • Acute intermittent porphyria. • Variegate porphyria. • Hereditary coproporphyria. • Non-acute porphyria: • Porphyria cutanea tarda. • Erythropoietic protoporphyria. • Congenital erythropoietic porphyria. Metabolic pathway • If enzymatic lesion occurs early then porphyrogins accumulate in tissues and fluids results in abdominal pain & neuropsychiatric symptoms. • If enzymatic lesion occurs later in pathway results in accumulation of porphyrin products causes photosensitivity & react with molecular oxygen followed by damage to lysosome and skin damage. Hepatic porphyrias • Increased plasma & urinary concentration of porphyrin precursors. Acute Intermittent Porphyria • Genetically autosomal dominant involving Uroporphyrogin I synthase. • Ecologically due to gonadal steroids and low Calorie diet. • Present with abdominal pain, neuro0psychiatricand cardiovasular symptom. Variants and types of hemoglobin (normally present at different ages) In embryo: • Gower 1 (ζ2ε2) • Gower 2 (α2ε2) • Hemoglobin Portland (ζ2γ2) In fetus: • Hemoglobin F (α2γ2) In adults: • Hemoglobin A (α2β2) - normal amount over 95% • • Hemoglobin A2 (α2δ2) - normal range of 1.5-3.5% Hemoglobin F (α2γ2) – normal range < 1% Variants and types of hemoglobin (variants which cause diseases) • • • • • • • Hemoglobin H (β4) - A variant form of hemoglobin, formed by a tetramer of β chains, which may be present in variants of α thalassemia Hemoglobin Barts (γ4) - A variant form of hemoglobin, formed by a tetramer of γ chains, which may be present in variants of α thalassemia. Hemoglobin S (α2βS2) - A variant form of hemoglobin found in people with sickle cell disease. There is a variation in the β-chain gene, causing a change in the properties of hemoglobin which results in sickling of red blood cells. Hemoglobin C (α2βC2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia. Hemoglobin E(α2βE2) - Another variant due to a variation in the β-chain gene. This variant causes a mild chronic hemolytic anemia Hemoglobin AS - A heterozygous form causing Sickle cell trait with one adult gene and one sickle cell disease gene Hemoglobin SC disease - Another heterozygous form with one sickle gene and another encoding Hemoglobin C Fetal hemoglobin (HbF) • HbF is a tetramer consisting of teo alpha chains identical to those found in HbA plus two gamma chains Synthesis during development: • In the first few weeks after conception,embryonic hemoglobin (Hb Gower 1 )composed of two zeta and two epsilon chains is synthesized by embryonic yolk sac. • Within few weeks,fetal liver begins to synthesize HbF in developing bone marrow • HbF is major hemoglobin found in fetus and newborn,accounting for 60% of total Hb in red cells during the last months of fetal life. • HbA synthesis starts in 8th month of pregnancy and gradually replaces HbF Binding of 2,3-BPG to HbF: • Under physiological conditions, HbF has higher affinity for oxygen than does HbA • This is because of result of HbF’s weak binding to 2,3-BPG • Because 2,3- BPG serves to reduce the affinity of HB for oxygen,the weak interaction between2,3-BPG and HbF results in higher oxygen affinity for HbF relative to HbA • The higher oxygen affinity of HbF facilitates the transfer of oxygen from the maternal circulation across placenta to the red cells of the fetus Hemoglobin A2 (HbA2) • A minor component of normal adult hemoglobin • First appears 12 weeks after birth • Constitutes 1.5 – 3.5 % of the total adult hemoglobin • Composed of two alpha and two delta globin chains Hemoglobin A1c • Under physiological conditions . HbA is slowly and non-enzymatically glycosylated and the extent depends on the plasma concentration of a particular hexose • The most abundant form of glycosylated hemoglobin is HbA1c • • • It has glucose residues attached predominantly to the NH2 groups of the N terminal valines of the beta globin chains Increased amounts of HbA1c are found in the red cells of patients with diabetes mellitus as they have got increased glucose concentrations in their plasma Very good indicator of the Plasma glucose control over a past 3 month interval – indicates patient’s compliance with respect to medication intake and the control of diet