Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
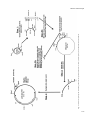
Carcinogenesis vol.18 no.7 pp.1311–1318, 1997 Site- and strand-specific mismatch repair of human H-ras genomic DNA in a mammalian cell line Loretta Arcangeli1,2, Josephine Simonetti1, Catherine Pongratz1 and Kandace J.Williams1,3 1Biomedical Program and Department of Biological Sciences, University of Alaska, Anchorage, Alaska 99508, USA 2Present address: Hopkins Marine Station, Stanford University, Oceanview Boulevard, Pacific Grove, CA 93950, USA 3To whom correspondence should be addressed Defective mismatch repair has recently been implicated as the major contributor towards the mutator phenotype observed in tumour cell lines derived from patients diagnosed with hereditary non-polyposis colon cancer (HNPCC). Cell lines from other cancer-prone syndromes, such as xeroderma pigmentosum, have been found to be defective in nucleotide excision repair of damaged bases. Some genetic complementation groups are defective specifically in transcription-coupled excision repair, although this type of repair defect has not been associated with cancer proneness. Mechanisms contributing to the high incidence of activating point mutations in oncogenes (such as H-ras codon 12) are not understood. It is possible that novel mechanisms of misrepair or misreplication occur at these sites in addition to the above DNA repair mechanisms. In this study, we have compared the rate of strand-directed mismatch repair of four mispairs (G:A, A:C, T:C and G:T) at the H-ras codon 12, middle G:C position. Our results indicate that, although this location is not a ‘hot spot’ for bacterial mismatch repair, it is a ‘hot spot’ for decreased repair of specific mismatched bases within NIH 3T3 cells. NIH 3T3, unlike Escherichia coli, have an extremely low repair rate of the G:A mispair (35%), as well as the A:C mispair (58%) at this location. NIH 3T3 also have a moderately low repair rate of the T:C mispair (80%) at the codon 12 location. Conversely, NIH 3T3 repair of G:T (100%) is comparable to E.coli repair (94%) of this mismatch. These results demonstrate that a mismatch containing an incorrect adenine on either strand at the H-ras codon 12 middle base pair location is most likely to undergo a mutational event in NIH 3T3 cells. Conversely, a mismatch containing an incorrect thymine in the transcribed strand is least likely to undergo a mutational event. Introduction The three primary gene products required for strand-specificity of mismatch repair in Escherichia coli, MutS, MutL and MutH, have been well described (1). These three enzymes work in a coordinated fashion to make a single-strand nick in the new strand of unmethylated DNA opposite the nearest methylated d(GATC) site, leading to either a 39 → 59 or 59 → 39 gap excision ‘long patch’ repair process to replace the incorrect *Abbreviations: HNPCC, hereditary non-polyposis colon cancer; RER1, replication errors; bp, base pair; ds, double strand. © Oxford University Press base. A series of recent studies have resulted in the identification of several human genetic homologues of bacterial DNA mismatch repair enzymes, such as hMSH2, hMLH1, hPMS1, hPMS2 and GTBP, although the exact biochemical function of each gene product has yet to be identified. Available evidence suggests that both the MSH2-GTBP (hMutSα) heterodimer and the MLH1-PMS2 heterodimer (hMutLα) can act as mismatch recognition complexes (1). Recent in vitro experiments have indicated that at least seven different biochemical activities are required for mammalian mismatch repair (2). Mechanisms of ‘new’ strand discrimination prior to mismatch repair within mammalian cells have not yet been elucidated. However, experiments using E.coli or human cell extracts have demonstrated strand-specificity of mismatch repair by the creation of a pre-existing nick on the same strand of DNA as the incorrect base, located either 59 or 39 from the mismatch (3–6). Within mammalian cells, there is more than one repair system for G:T or G:U mispairs putatively arising from endogenous 5-methylcytosine or cytosine deamination events, respectively (7,8). One well-described repair mechanism for the G:T mispair is a base excision repair process using a specific thymine glycosylase that does not require stranddiscrimination (9). In addition, it has recently been reported that the GTBP polypeptide of the hMutSα complex appears to bind preferentially with G:T mispairs (10). One more mismatch repair complex, hMutLα (composed of hMLH1 and hPMS2) can restore strand-discriminatory repair activity for all eight mismatches in nuclear extracts of a human tumour cell line (6). It is now evident that all combinations of mismatches can be repaired in mammalian cells; however, G:T mispairs appear to be corrected with higher efficiency than other mispairs, perhaps due to a wider repertoire of repair mechanisms (1,11). The majority of patients who have been diagnosed with hereditary non-polyposis colon cancer (HNPCC*) have germline mutations in one or more of the human mismatch repair homologues. Studies using tumour cell lines developed from these patients have demonstrated a distinct mutator phenotype that appears to have a substantial increase in replication errors (RER1) and therefore, may be involved in subsequent tumour progression. RER1 cells commonly exhibit microsatellite instability, an increase in HPRT gene mutability, insertiondeletion mutation events and tolerance to alkylation base damage. Furthermore, extracts from these cells have an inability to perform nick-directed mismatch correction (2). DNA mismatch repair in these cells is blocked prior to the gap excision stage. In addition, human mismatch repair deficiencies have recently been implicated in deficiencies of transcriptioncoupled nucleotide excision repair of damaged nucleotides (12). Progress in understanding the link between mismatch repair, the mutator phenotype and cancer in human cells has been phenomenally rapid. Issues yet to be resolved include biochemical interactions between mismatch repair complexes and DNA, and differential repair of specific mismatched bases in sensitive 1311 L.Arcangeli et al. genomic locations. Virtually all mismatch repair information obtained to date has resulted from experiments using synthetic or bacterial DNA sequences and bacterial assays or mammalian cell extracts. Therefore, information about the differential repair of specific mammalian genetic sequences within mammalian cells is essentially unknown. Oncogenes and tumour suppressor genes containing distinct ‘hot spots’ of mutation are of high interest in regard to accuracy of DNA repair processes as well as fidelity of DNA replication. Activating mutations in these genes consistently found in human tumours may be due partly to a genetic defect in one of the DNA repair pathways, such as mismatch repair or nucleotide excision repair (13). However, to account for the high specificity and frequency of mutations at these hot spots, it is probable that a yet to be identified mechanism of misreplication or misrepair plays an additional role at these sensitive locations in the genome. Clearly, etiology of cellular transformation has yet to be elucidated in regard to the high mutation rate in these susceptible genomic locations. The subject of this work has been to examine differences in both rate and accuracy of in vivo site-, strand- and basespecific mismatch repair located at a well known ‘hot spot’ in the human H-ras oncogene. The objectives of this research have been to determine differences, if any, in the frequency and accuracy of mammalian mismatch repair proficiency at a sensitive oncogenic location (H-ras codon 12, middle nucleotide) depending on type and orientation of each mismatched base. Mismatch repair rates have also been compared between mammalian and bacterial organisms to establish a baseline understanding of differences in mismatch repair mechanisms that may exist between these diverse species. We use a unique, highly sensitive assay previously developed in this laboratory for studying mammalian cellular repair of DNA damage at a relevant location in regard to cellular transformation (14). Materials and methods Enzymes T4 DNA ligase, calf intestinal alkaline phosphatase (CIP), KpnI, XhoI and BfrI were purchased from Boehringer Mannheim Biochemicals. T4 polynucleotide kinase was purchased from Promega. Replitherm DNA polymerase was purchased from Epicentre Technologies. BamHI and HpaII were purchased from New England Biolabs. HindIII was purchased from United States Biochemical. Proteinase K was purchased from Sigma Chemical Co. Shrimp alkaline phosphatase I (SAPI), exonuclease I (ExoI) and thermosequenase were purchased from Amersham. Cells, plasmids, oligonucleotides and other reagents NIH 3T3 cells were obtained from American Type Culture Collection (ATCC). The construction of plasmids p220.pbc and p220.pbc1H/B has been described previously (14; Figure 1). M13mp18, M13mp19, pUC19, DH5α competent E.coli, LipofectAMINE, and Opti-MEM were purchased from Life Technologies, Inc. E.coli strain NR9161 (-mutL) was a kind gift from Roel Schapper (NIEHS, Research Triangle Park, NC). All synthetic oligonucleotides were purchased from Operon Technologies, Inc. Radioactively labelled nucleotides were purchased from Dupont–New England Nuclear. Agarose for electrophoretic separation and purification of DNA was purchased from FMC Bioproducts. Gene Clean II was purchased from Bio 101, Inc. Dulbecco’s Modified Eagle’s Medium (DMEM; 4.5 g/l glucose) and bovine calf serum were purchased from Hyclone Laboratories, Inc. Hygromycin B was purchased from Calbiochem Biochemicals. All other reagents were purchased from Sigma Chemical Company unless otherwise noted. Formation of p220.pbc1H/B containing H-ras codon 12 site-specific mismatch A 2 kb BamHI–KpnI H-ras segment from p220.pbc1H/B (containing codon 12) was ligated into the polylinker region of pUC19 (prasBK2.0), M13mp18 (M13ras18.9) and M13mp19 (M13ras19.1), as described previously (14). The plasmid prasBK2.0 was digested with AflIII and PvuI to release a 2.5 kb fragment containing the inserted H-ras segment. The purified 2.5 kb DNA 1312 fragment was then digested with HindIII and BfrI to remove the original 30base pair (bp) segment containing codon 12 of H-ras (Figure 1, step 1). Mismatch oligonucleotides complementary to the 30 bp region spanning codon 12 (middle dG replaced with dT or dA for non-transcribed strand mismatch oligomer, or middle dC replaced with dT or dA for transcribed strand mismatch oligomer) were phosphorylated and purified by G-50 Sephadex column as described previously (14). Generation of specific mismatched heteroduplex DNA (HD DNA) was made according to choice of mismatch 30-mer oligo and single-strand (ss) M13ras complementary DNA. Use of ss M13ras18.9 results in a 30-bp double-strand (ds) region containing either dT or dA mismatched codon 12 middle nucleotide in the coding strand of H-ras (non-transcribed strand). Inversely, use of ss M13ras19.1 results in a 30-bp ds region containing either dT or dA mismatched codon 12 middle nucleotide in the non-coding strand of H-ras (transcribed strand). The phosphorylated mismatch oligo was annealed with the respective ss M13ras DNA at a 50:1 molar ratio and a total DNA concentration of 50 ng/µl in TE (10 mM Tris, pH 8.0, 1 mM EDTA) at 65°C for 15 min, resulting in an M13ras with a 30 bp mismatch insert on one strand (Figure 1, step 2A). H-ras fragments digested and purified from prasBK2.0 complementary to the regions flanking the 30-bp mismatch region were heatdenatured at 100°C for 8 min and annealed with the ss M13/mismatch oligo HD DNA at a 1:2 molar ratio at 65°C for 30 min in annealing buffer (40 mM Tris, pH 7.5, 20 mM MgCl2, 50 mM NaCl) (Figure 1, step 2B). This partially ds M13ras molecule was cleaved with XhoI and KpnI to produce a 1.8 kb ds fragment (Figure 1, step 3), that was then purified, gently dephosphorylated (to discourage 1.8 kb concatemer ligation; 0.01 U CIP per pmol of 59 termini for 1 h at 37°C) and ligated to the 13.5 kb XhoI–KpnI vector portion of p220.pbc at a 4:1 molar ratio, and additional phosphorylated mismatch oligo at a 200:1 molar ratio, at 16°C overnight. This final step produces the p220.pbc1H/B plasmid, containing a site- and strand-specific mismatch at Hras codon 12, middle base pair (Figure 1, step 4). Figure 2 depicts a comparative NarI restriction digestion of a purified 1.8 kb XhoI/KpnI segment of control H-ras DNA and the same segment that has been obtained after preparation of site- and strand-specific mismatch DNA, as described above (Figure 1, steps 1–3). Although some of the NarI cleaved DNA segments apparently reflect missing NarI sites within intronic regions of the 2.0 kb segment of H-ras after cloning into the M13 bacteriophage, the two bands representing the distances between the NarI site at codons 10/11 (nucleotide no. 1693 of human genomic H-ras sequence) to the adjacent NarI site downstream of exon 1 (no. 1280), as well as to the KpnI site upstream of exon 1 (no. 1967), appear identical in size for both DNA segments, 413 bp and 274 bp, respectively, indicating that H-ras exon 1 (nos 1664–1774) is intact and the annealed mismatch 30-mer is in place within the site- and strand-specific mismatch DNA. Transfection, selection and analyses of human H-ras DNA from ampicillin resistant E.coli and hygromycin resistant NIH 3T3 cells Competent DH5α E.coli were transformed with mismatched plasmid DNA following the manufacturer’s protocol (Life Technologies, Inc.). NR9161 E.coli were made competent and transformed by the calcium chloride procedure (15). Bacteria were grown overnight at 37°C on LB agar plates containing 75 µg/ml carbenicillin. NIH 3T3 cells were grown in DMEM, 10% calf serum at 37°C, 5% CO2. Cells were seeded at 13106 per 100 mm plate in preparation for transfection experiments 16–18 h later. Plasmid DNA (50 ng per plate) was transfected into NIH 3T3 cells using LipofectAMINE reagent as described by the manufacturer (Life Technologies, Inc.). NIH 3T3 hygromycin resistant cells were subsequently selected by the addition of 125 U of hygromycin per millilitre of media, starting 36 h after transfection. At the end of 3 weeks, hygromycin resistant colonies on each plate were methanol (97%) fixed. DNA was purified from each E.coli and NIH 3T3 colony and amplified by PCR, yielding an amplified DNA product of 129 bp containing the entire exon 1 of human H-ras plus several human intronic nucleotides surrounding exon 1, as described previously (14). A 20-µl aliquot of the amplified DNA was then digested with HpaII restriction enzyme. Only PCR amplified DNA containing wild-type human H-ras codon 12 sequence (GGC) is cleaved by HpaII, yielding an 88 bp and a 41 bp band from the original 129 bp sequence (Figure 3A). All PCR amplified DNA not completely cleaved by HpaII was treated with the enzymes SAPI and Exol and cycle-sequenced, as described by the manufacturer (Amersham). All contaminating sequences, either from original p220.pbc used for the 13.5 kb vector DNA, or wild type transfection control DNA (codon 12; middle G:C), or from p220.T24 used for activated transfection control DNA (codon 12; middle T:A), were detected by observing sequences at codons 6 and 15, both of which have been altered to create either a unique HindIII site (codon 6) or BfrI site (codon 15) in the final p220.pbc1H/B plasmid containing the site-specific mismatch (14). Any sequences not containing the altered sequences at codons 6 (CTG → CTT) and 15 (GGC → CTT) were discarded. Fig. 1. Construction of p220.pbc1H/B containing strand- and site-specific mismatch at H-ras codon 12, middle position, as described in Materials and methods. Defective mismatch repair 1313 L.Arcangeli et al. Results Although it is not clear why this large plasmid will not transfect into cells efficiently if it is not in the ds circular form, it does serve as a control to exclude simple ‘gap repair’ results from true mismatch repair results. To determine experimentally if this method would, indeed, bias mismatch repair to the incorrect base by nicked-strand specificity, each mismatch experiment was performed by transfecting aliquots of the same mismatched ligation products into both E.coli [DH5α as mismatch repair competent strain; NR9161 (-mutL) as mismatch repair defective strain] and NIH 3T3 cells. Table I demonstrates the overall high rate and high fidelity of correct mismatch repair (to G:C) at this site for all four of the mismatches tested by mismatch repair competent E.coli (DH5α). Table I also illustrates 100% negative mismatch repair by mismatch repair defective E.coli (NR9161). Results from NR9161 experiments indicate the high purity of the sitespecific mismatch ligation products used in these experiments, although this may be due, in part, to cellular selectivity for ds (non-gapped) circular plasmids. Table II contains results of NIH 3T3 mismatch repair of aliquots of the same ligation products for each mismatch. All experiments were repeated with separately prepared mixtures of ligation products to ensure reproducibility of results. This assay system therefore appears to represent a true comparison of mismatch repair capabilities of this oncogenic ‘hot spot’ by a mammalian cell line and by a prokaryotic organism for each of the four mismatches tested. Fidelity of cellular mismatch repair at a ‘hot spot’ of oncogenic activation has been investigated using an Epstein–Barr virus (EBV) plasmid vector containing genomic human H-ras DNA (14). Specific base pair mismatches are located at the middle nucleotide position of codon 12: either dG of the coding strand or dC of the transcribed strand has been replaced with dA or dT. Site- and strand-specific mismatches are constructed by annealing complementary 30 base oligomers and surrounding H-ras fragments to a ss M13ras plasmid to produce heteroduplex DNA that is then cleaved by restriction digestion and religated into the remaining portion of the original plasmid vector (Figure 1). This methodology does not facilitate hemimethylation d(GATC) directed mismatch repair by E.coli. It is also unlikely that the initiating mechanism of mammalian strand-directed mismatch repair is facilitated. However, this procedure does produce a ‘pre-ligation’ total of four ‘nicks’ on the DNA strand containing the incorrect mismatched nucleotide (each end of the 1.8 kb ds fragment plus each end of the ss 30-mer mismatch oligo) versus two ‘nicks’ on the opposite DNA strand (each end of the ds 1.8 kb fragment only) during preparation of mismatched DNA (Figure 1). The highly unlikely event of 100% in vitro ligation efficiency would require DNA ligase to ligate each end of the ss 30-mer oligonucleotide containing the incorrect base as well as both strands of each end of the ds 1.8 kb fragment of DNA into the remaining 13.5 kb portion of p220.pbc, for a total of six ligations per plasmid (Figure 1, step 4). Therefore, this protocol was determined to be appropriate for biasing the selection for plasmids containing unligated ‘nicks’ on either end of the ss 30-mer containing the incorrect base and/or on the same DNA strand on either end of the 1.8 kb insert. In addition, we have repeatedly observed that this 15.3 kb plasmid has an ~50-fold higher transfection efficiency in NIH 3T3 cells in the ds circular form than when it is linear or gapped (missing 30mer oligo). We have been unable to transform E.coli successfully with this plasmid when it is in the linear or gapped form. Mismatch repair analyses Figure 3A illustrates HpaII restriction digestion and sequencing results of ‘activated’ and ‘wild-type’ H-ras codon 12 PCR amplified DNA. The PCR amplified 129 bp DNA band that did not cut in the presence of HpaII (‘activated’) was sequenced to reveal a GGC → GTC activation mutation at codon 12. The PCR amplified DNA that was cleaved by HpaII (‘wildtype’) to yield an 88 bp and 41 bp band was sequenced to reveal the expected GGC sequence at codon 12. Figure 3B illustrates the results of a typical experiment in which E.coli were transformed with p220.pbc1H/B containing a codon 12, middle G:A mismatch. H-ras DNA from individual carbenicillin resistant colonies was PCR amplified and cleaved with HpaII. Lanes 1, 3, 4, 7 and 8 do not appear to be cleaved by HpaII. Sequencing the amplified DNA from these colonies reveals a GGC → GTC transversion mutation. Lanes 5 and 12 contain amplified DNA that appears to be mostly or completely cleaved by HpaII. Subsequent sequencing revealed the expected GGC wild type sequence. The remaining lanes in Figure 3B contain DNA that is only partially cleaved by HpaII. Subsequent sequencing revealed a mixture of GGC and GTC at codon 12. Figure 3C demonstrates sequencing results from a typical mismatch experiment in which NIH 3T3 cells were transfected with the same plasmid DNA preparations containing a G:A mismatch. Sequences 3, 4, 5, 6, 7, 9 and 10 have been corrected to wild-type (GGC). The remaining five sequences are G/T mixtures at the middle nucleotide of codon 12, similar to lanes 2, 6, 9, 10 and 11 in Figure 3B. This could indicate that either DNA replication has occurred before mismatch correction or the presence of more than one plasmid per cell, with each mismatch repaired differently. Either situation could result in a mixture of wild-type and activated H-ras DNA. However, because subsequent results during NIH 3T3 experiments revealed a wide range of frequency of mixtures (0–65%), depending on the mismatch, it is more probable that DNA Fig. 2. NarI restriction cleavage and agarose electrophoresis to compare control ds 1.8 kb XhoI/KpnI DNA fragments with G:A mismatch ds 1.8 kb XhoI/KpnI DNA fragments. The two DNA bands, 413 bp and 274 bp, represent the distance between NarI recognition sites nos 1280 and 1693 (codons 10/11 location) and between NarI recognition site no. 1693 and KpnI site no. 1967. Exon 1 of H-ras spans nos 1664–1774. 1 kb ladder is shown on the left for size determinations. Sequence diagram on right not drawn to scale. 1314 Defective mismatch repair Fig. 3. PCR amplification and HpaII restriction digestion of H-ras mismatch plasmid DNA after replication in NIH 3T3 cells and E.coli. (A) HpaII cleavage and sequencing results of PCR amplified ‘activated’ (codon 12 GTC) and ‘wild-type’ (codon 12 GGC) control plasmid DNA after replication in NIH 3T3 cells. (B) HpaII cleavage and sequencing results of PCR amplified H-ras G:A mismatch plasmid DNA after replication in E.coli. (C) Sequencing results of PCR amplified H-ras G:A mismatch plasmid DNA after replication in NIH 3T3 cells. replication has occurred before repair in these mixtures, and therefore competent cellular mismatch repair is dependent on the specific type of mismatch at this location (Table II). Table I (E.coli) and Table II (NIH 3T3) are compiled results of several experiments examining frequency of correct repair back to G:C, incorrect repair to A:T or T:A or putative ‘unrepair’ by the presence of mixtures of G:C and A:T or T:A for four different mismatches (G:A, A:C, T:C, G:T) at the human H-ras codon 12 ‘hot spot’ of mutation. Table I contains results from a total of 306 DH5α colonies and 23 NR9161 (-mutL) colonies. Of the four mismatches examined after replication in DH5α, G:A mismatches were repaired correctly back to G:C at the lowest rate of 87% (78/ 90) and repaired incorrectly at the highest rate of 8% (7/90). 1315 L.Arcangeli et al. Table I. E.coli mismatch repair of human H-ras codon 12 ‘hot spot’ Repaired to Mismatch (DH5α)a Correctly repaired (Mismatch → G:C) (Total assayed) Incorrectly repaired (Mismatch → A:T or T:A) (Total assayed) Unrepaired (Mismatch → Mixturesb) (Total assayed) (NR9161)c Unrepaired (Mismatch → Mixturesb) (Total assayed) G:A A:C T:C G:T 87% (78/90) 97% (84/87) 100% (69/69) 94% (56/60) 8% (7/90) 2% (2/87) 0% (0/69) 3% (2/60) 5% (5/90) 1% (1/87) 0% (0/69) 3% (2/60) n.d. n.d. 100% (7/7) 100% (16/16) aDH5α are wild-type E.coli bMixtures are G:C and A:T cNR9161 with no mismatch repair defect. or T:A, depending on mismatch. are mismatch repair defective E.coli (-mutL). Table II. NIH 3T3 mismatch repair of human H-ras codon 12 ‘hot spot’ Repaired to Correctly repaired (Mismatch → G:C) (Total assayed) Incorrectly repaired (Mismatch → A:T or T:A) (Total assayed) Unrepaired (Mismatch → mixturesb) (Total assayed) Mismatch G:A A:C T:C G:T Gappeda 35% (24/69) 58% (25/43) 80% (36/45) 100% (67/67) 100% (23/23) 0% (0/69) 0% (0/43) 0% (0/45) 0% (0/67) 0% (0/23) 65% (45/69) 42% (18/43) 20% (9/45) 0% (0/67) 0% (0/23) aGapped results from transfection of complete circular p220.pbc1H/B plasmid without 30-mer mismatch oligomer. bMixtures are G:C and A:T or T:A, depending on mismatch. G:A mismatches also had the highest rate at 5% (5/90) of unrepaired mismatch before replication (resulting in a mixture of wild-type and mutated end product). The other three mismatches examined at this site (A:C, T:C and G:T) were repaired correctly back to G:C by DH5α at overall high rates of 97% (84/87), 100% (69/69) and 94% (56/60), respectively. Although only T:C and G:T mismatches were used to transform NR9161 (-mutL), 100% of the colonies examined contained unrepaired H-ras DNA as evidenced by mixtures of wild-type and mutated sequences at the codon 12 location. Table II contains results of PCR amplified human H-ras DNA from a total of 247 hygromycin resistant NIH 3T3 colonies. Only 35% (24/69) of G:A mismatches were repaired correctly back to G:C with the remaining unrepaired. Only 58% (25/43) of A:C mismatches were repaired correctly back to G:C, with the remaining unrepaired. A total of 80% (36/45) of the T:C mismatches were repaired correctly back to G:C, again with the remaining unrepaired. A total of 100% (69/69) of G:T mismatches were repaired correctly back to G:C. Also, 100% (23/23) of gapped plasmids (circular 1316 p220.pbc1H/B without 30-mer mismatch oligomer) were gapfilled/repaired correctly to produce a G:C base pair at codon 12. An overall comparison of Tables I and II demonstrates that for specific mismatches at this location, NIH 3T3 cells are considerably less capable of performing mismatch repair than mismatch repair competent DH5α. G:A mismatch repair occurs with lowest fidelity of all four mismatches examined in both organisms, although correct G:A mismatch repair to G:C in DH5α (87%) is still significantly higher than in NIH 3T3 (35%). Interestingly, G:A and A:C mismatch repair in NIH 3T3 (35 and 58%, respectively) are the lowest repair rates of all four mismatches at this location, with T:C (80%) and G:T (100%) repair rates increasingly improved, respectively. The repair rates of A:C (58%) and T:C (80%) are somewhat in contrast to our previous report of A:C (87.5%) and T:C (67%) repair rates, in which we used a different method to produce the site- and strand-specific mismatch (14). It is not clear what may have caused this difference in results, perhaps there are differences in distribution of residual ‘nicks’ after construction by each method. Current experiments indicate that NIH 3T3 cells do not appear to have any increased efficiency of mismatch repair of transcriptional versus coding strand at this site (G:T . T:C .. A:C .. G:A), but NIH 3T3 cells do appear to have significantly decreased repair efficiency of adenine (35%, 58%) as compared to thymine (80%, 100%), regardless of which DNA strand contains the incorrect adenine. Interestingly, in contrast to DH5α, when mismatch repair does occur in NIH 3T3, it is consistently accurate repair back to G:C, as evidenced by 0% mutation rates (incorrectly repaired) for all mismatches tested in Table II (provided that mixtures are truly indicative of unrepaired plasmids). Discussion The experimental system described in this paper has been developed to study the frequency and accuracy of mammalian DNA repair when a strand- and site-specific lesion is introduced at a major ‘hot spot’ of mutation in the human H-ras oncogene. The mammalian expression vector used for these studies contains the entire human H-ras genomic sequence and a hygromycin resistance gene for selection of mammalian clones expressing this plasmid. In addition, this EBV vector replicates synchronously with the cell cycle, maintains a low spontaneous mutation rate, and a low plasmid copy number per cell (14). Our methodology permits site-specific incorrect base insertion either on the transcribed or non-transcribed (coding) strand. One limitation to our approach, however, has been our inability to create strand-specific nicks as clearly defined as in vitro systems currently in the literature (3–6,10,16). Instead, we have relied on a strand-biasing approach that involves placing four of the initial six nicks on the same DNA strand as the incorrect base during plasmid preparation. Subsequent in vitro ligation conditions are unlikely to approach 100% efficiency, therefore an unligated nick in the same strand as the incorrect base is highly favoured. This approach appears feasible when observing results from DH5α transformation experiments from several different plasmid and mismatch preparations (Table I). Also, results obtained in this laboratory agree with those of other investigators in regard to the high rate and fidelity of strand-directed DH5α mismatch repair (17,18). In addition, two separate mismatch experiments using mismatch deficient NR9161 E.coli indicate that each plasmid preparation contains the mismatch at the time of transformation, Defective mismatch repair as amplified H-ras DNA from each of these colonies remains unrepaired. In NIH 3T3 cells, however, aliquots of the same Hras codon 12 site- and strand-specific mismatch plasmid preparations do not appear to be repaired with a similarly high rate as DH5α, using this experimental system (Table II), with the exception of G:T → G:C (100%). Table II also indicates a wide variation in rate of specific mismatch repair observed with NIH 3T3 cells as compared to DH5α. There could be several reasons for these results. Currently, mammalian cellular DNA repair mechanisms are not as well understood as E.coli DNA repair. Therefore, while the strand-nick bias conditions in these experiments appear sufficient for DH5α strand-directed mismatch repair, the same conditions may not be sufficient for NIH 3T3 strand-directed mismatch repair. We are currently experimenting with different methods of constructing a more specifically defined ‘nicked’ mismatch-containing plasmid to resolve this issue. Interestingly, however, our current experiments indicate that when mismatch repair does occur in NIH 3T3 cells, fidelity of correct repair back to G:C is a consistent event, as opposed to incorrect repair resulting in a mutation (Table II). Therefore, regardless of the rate of mismatch repair, strand-directed correct mismatch repair appears to play a significant role in NIH 3T3. Replication errors that are not corrected by proof-reading processes during DNA replication are thought to be corrected by strand-discriminating mechanisms of mismatch repair at a stage of the cell cycle other than S phase. Cell cycle synchronization studies in progress in this laboratory should define precisely differences in specific mismatch repair rates at this oncogenic location, during different discrete stages of the mammalian cell cycle. Additionally, NIH 3T3 cells have a high rate of spontaneous transformation to the malignant phenotype (19). Perhaps these cells have some, as yet, unidentified deficiency in specific mismatched base repair, contributing to their mutator phenotype. To test this possibility, we are currently assessing different mammalian cell lines for efficiency and fidelity of mismatch repair. Codon 12 of H-ras is a well-known hot spot of mutagenesis in human tumors and animal model studies (20–22). Mechanisms (or lack thereof) contributing to the increased rate of mutation at this, and other precisely targeted activating locations, are not yet known. One hypothesis to explain such precise mutagenic targeting is that specific types of DNA damage occur more frequently at certain sites in the genome (21). A second hypothesis is that there is random DNA damage in the genome with decreased repair of damage at specific sites (13,22). A third hypothesis is that cells with certain activating mutations are selected for increased survival (23). It is possible that none of these hypotheses are mutually exclusive. We are in the process of comparing frequency and fidelity of mismatch repair at similar codons near codon 12 in H-ras that are not frequent sites of mutation to determine if the specific types of mismatch repair observed in these studies are decreased precisely because they occur at the codon 12 location of H-ras. Tables I and II indicate that both DH5α and NIH 3T3 have increased difficulties specifically with the G:A mismatch at this location, supporting results of other investigators that this is one of the most difficult mismatches for all organisms to repair (1,24). This may be because G:A is not easily recognized as a mismatch, or because of the diverse physical conformations this mismatch can assume within the DNA molecule, perhaps confusing enzymatic recognition (25). A:C mismatches have been noted for high fidelity and rate of correct mismatch repair in other systems (11,16,24,25). Our current results, using NIH 3T3 cells, do not agree with others for a high rate of repair of this particular mismatch at this oncogenic location (58%). Most strikingly, the same A:C mismatch plasmid preparation is repaired with high consistency by DH5α (97%). The mismatch at this site that is most efficiently and accurately repaired by NIH 3T3 is the G:T mismatch (100%), which is in agreement with the results of other investigators (1,10,11,26). This is probably a frequently occurring mismatch at this heavy CpG region of DNA, due to endogenous deamination of 5methylcytosine, and therefore might have more than one enzyme complex available for accurate repair (9,10). Likewise, the T:C mismatch has been previously demonstrated also to be repaired by a specific G:T mismatch enzyme, thymine DNA glycosylase (9), in addition to strand-directed mismatch repair, which may explain the higher rate of repair of this mismatch in these experiments. Perhaps the most interesting aspect of NIH 3T3 mismatch repair in this study is the significant difference in repair rates of mismatches containing an incorrect adenine (G:A and A:C) versus thymine (T:C and G:T), regardless of strand placement of the incorrect base. Clearly, a mismatch containing an incorrect adenine at this site is most likely to undergo a mutational event. Cell cycle synchronization experiments, mismatch repair at other site-specific locations in exon 1 of H-ras, and experiments examining the effect of a specific strand-break either 39 or 59 of each mismatch, currently ongoing in this laboratory, should prove interesting to compare with the inefficiently repaired mismatches by NIH 3T3 in this study. Similar mismatch studies using other cell lines are also under way. Results in Tables I and II argue that although codon 12, middle G:C does not appear to be a strong ‘hot spot’ for infidelity of bacterial mismatch repair, this location does appear to be a ‘hot spot’ for lack of repair by NIH 3T3 cells containing a G:A or A:C mispair, and possibly a T:C mispair. Each mismatch, not correctly repaired, results in a T:A or A:T ‘activating’ mutation frequently observed at this site in human tumours. The results of this study clearly demonstrate that it is possible to obtain information about mammalian mismatch repair in vivo at a relevant genomic location involved with cell transformation. Future results using this system should contribute significantly towards a clearer understanding of mechanisms of mutation and initiation of carcinogenesis. Acknowledgement This work was supported by NIH grant CA-57495 from the National Cancer Institute. References 1. Modrich,P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination and cancer biology. Ann. Rev. Biochem., 65, 101–133. 2. Modrich,P. (1995) Mismatch repair, genetic stability and tumour avoidance. Phil. Trans. Roy. Soc. Lond. B, 347, 89–95. 3. Umar,A., Boyer,J.C. and Kunkel,T.A. (1994) DNA loop repair by human cell extracts. Science, 266, 814–816. 4. Risinger,J.I., Umar,A., Barrett,J.C. and Kunkel,T.A. (1995) A hPMS2 mutant cell line is defective in strand-specific mismatch repair. J. Biol. Chem., 270, 18183–18186. 1317 L.Arcangeli et al. 5. Drummond,J.T., Li,G.-M., Longley,M.J. and Modrich,P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science, 268, 1909–1912. 6. Li, G.-M. and Modrich,P. (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl Acad. Sci. USA, 92, 1950–1954. 7. Stephenson,C. and Karran,P. (1989) Selective binding to DNA base pair mismatches by proteins from human cells. J. Biol. Chem., 264, 21177–21182. 8. Griffin,S., Branch,P., Xu,Y.-Z. and Karran,P. (1994) DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA methylation damage. Biochemistry, 33, 4787–4793. 9. Neddermann,P. and Jiricny,J. (1993) The purification of a mismatchspecific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem., 268, 21218–21224. 10. Palombo,F., Gallinari,P., Iaccarino,I., Lettieri,T., Hughes,M., D’Arrigo,A., Truong,O., Hsuan,J.J. and Jiricny,J. (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science, 268, 1912–1914. 11. Brown,T.C. and Jiricny,J. (1988) Different base/base mispairs are corrected with different efficiencies and specificities in monkey kidney cells. Cell, 54, 705–711. 12. Mellon,I., Rajpal,D.K., Koi,M., Boland,C.R. and Champe,G.N. (1996) Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science, 272, 557–560. 13. Bohr,V.A. (1995) DNA repair fine structure and its relations to genomic instability. Carcinogenesis, 16, 2885–2892. 14. Arcangeli,L. and Williams,K.J. (1995) Mammalian assay for site-specific DNA damage processing using the human H-ras proto-oncogene. Nucl. Acids Res., 23, 2269–2275. 15. Sambrook,J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 16. Fang,W.-H. and Modrich,P. (1993) Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem., 268, 11838–11844. 17. Langle-Rouault,F., Maenhaut-Michel,G. and Radman,M. (1987) GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch repair. EMBO J., 6, 1121–1127. 18. Lahue,R.S., Su,S.-S. and Modrich,P. (1987) Requirement for d(GATC) sequences in Escherichia coli mutHLS mismatch correction. Proc. Natl Acad. Sci. USA, 84, 1482–1486. 19. Rubin,H. and Xu,K. (1989) Evidence for the progressive and adaptive nature of spontaneous transformation in the NIH 3T3 cell line. Proc. Natl Acad. Sci. USA, 86, 1860–1864. 20. Barbacid,M. (1987) ras genes. Ann. Rev. Biochem., 56, 779–827. 21. Bos,J.L. (1989) ras oncogenes in human cancer: a review. Cancer Res., 49, 4682–4689. 22. Balmain,A. and Brown,K. (1988) Oncogene activation in chemical carcinogenesis. Adv. Cancer Res., 51, 147–182. 23. Guerrero,I. and Pellicer,A. (1987) Mutational activation of oncogenes in animal model systems of carcinogenesis. Mutat. Res., 185, 293–308. 24. Brown,T.C. and Jiricny,J. (1989) Repair of base-base mismatches in simian and human cells. Genome, 31, 578–582. 25. Brown,T. (1995) Mismatches and mutagenic lesions in nucleic acids. Aldrichimica Acta, 28, 15–20. 26. Brown,T.C. and Jiricny,J. (1987) A specific mismatch repair event protects mammalian cells from loss of 5-methylcytosine. Cell, 50, 945–950. Received on July 22, 1996; revised on March 14, 1997; accepted on March 17, 1997 1318