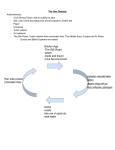
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
DNA vaccination wikipedia , lookup
Non-coding RNA wikipedia , lookup
Non-coding DNA wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
History of RNA biology wikipedia , lookup
Primary transcript wikipedia , lookup
Protein moonlighting wikipedia , lookup
Metagenomics wikipedia , lookup
Frameshift mutation wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Helitron (biology) wikipedia , lookup
Expanded genetic code wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
Sequence alignment wikipedia , lookup
BioMath Spider Silk: Examining Biological Sequences Student Edition Funded by the National Science Foundation, Proposal No. ESI-06-28091 This material was prepared with the support of the National Science Foundation. However, any opinions, findings, conclusions, and/or recommendations herein are those of the authors and do not necessarily reflect the views of the NSF. At the time of publishing, all included URLs were checked and active. We make every effort to make sure all links stay active, but we cannot make any guaranties that they will remain so. If you find a URL that is inactive, please inform us at [email protected]. DIMACS Published by COMAP, Inc. in conjunction with DIMACS, Rutgers University. ©2015 COMAP, Inc. Printed in the U.S.A. COMAP, Inc. 175 Middlesex Turnpike, Suite 3B Bedford, MA 01730 www.comap.com ISBN: 1 933223 69 3 Front Cover Photograph: EPA GULF BREEZE LABORATORY, PATHO-BIOLOGY LAB. LINDA SHARP ASSISTANT This work is in the public domain in the United States because it is a work prepared by an officer or employee of the United States Government as part of that person’s official duties. Spider Silk: Examining Biological Sequences Stronger than steel and more elastic than rubber: spider silk is unsurpassed in its expandability, resistance to tearing, and toughness. Spider silk would be an ideal material for a large variety of medical and technical applications, and researchers are thus interested in learning the spiders’ secrets and imitating their technique.[1] This unit has a direct purpose and an indirect purpose. Its direct purpose is to explain some of the amazing properties of spider silk, how a mathematical algorithm called “sequence alignment” can be used to uncover some of its secrets, and how a computing environment can be employed to quickly implement this algorithm. Its secondary purpose is to show students that biology and mathematics are more interdependent now than ever before, and that mathematical skills will continue to grow in importance as an essential tool for biology research. Spiders and Silk Spiders are classified in the animal kingdom as shown below: • Phylum Arthropoda (which includes insects, arachnids, and crustaceans) • Class Arachnida (which includes scorpions, mites and ticks) • Order Araneae (which contains thousands of spider species) Spiders are found worldwide and most are predators of insects. As predators, spiders play an important ecological role in controlling insect populations. Spiders have a variety of methods for capturing prey. Some produce toxins that immobilize prey, some physically catch prey, and others build webs to trap their prey. The activities in this unit will deal with a few of the webbuilding species. Most species of spiders produce several types of silk, each having a specific purpose. These include constructing webs, capturing prey, assisting movement, and protecting eggs. The silk is a solid fiber composed of different proteins combined to provide the mechanical properties necessary for each function. The thread of protein forming the silk is released from an internal gland and passes through structures called “spinnerettes,” located on the abdomen, which remove moisture, and produce a solid fiber. Usually each species of spider produces several types of silk, each released by a different silk gland. In the most familiar type of spider, the orb-weaver, the web is a flat spiral anchored in several directions to a structure of some sort, perhaps a wall, a branch, or a leaf. Major ampullate or dragline silk makes up the axes of the web and anchors the web to a support. Minor ampullate silk is applied to the support in a spiral starting in the middle where the draglines intersect. It is attached to the dragline silk with a piriform silk that is glue-like. As the spiral increases in diameter, the silk changes from minor ampullate to flagelliform silk for the part of the web where insects are likely to impact. Flagelliform is much more elastic than minor ampullate, so the insect does not bounce off, but becomes ensnared with the forward and backward stretching of the web. Spider Silk Student 1 The strength and mechanical properties of spider silks are extraordinary. They have high tensile strength (are hard to break), are sticky and very elastic. Dragline silk is stronger than KEVLAR (used in bullet-proof vests) and the tendons in human joints. Flagelliform is also stronger than KEVLAR, 40 times more elastic than tendons, and one-third as elastic as rubber. The silks are also insoluble in water; webs stand up to rain storms and dew quite well. In fact, dragline silk shrinks when wet to about 50% of its dry length. Because of these properties, artificially produced spider silk could be used to produce such things as artificial tendons, sutures used in surgery, lightweight bulletproof vests, and wear-resistant clothes. Unfortunately, spiders are not social creatures, so it is not possible to have spiders live in colonies in order to harvest their silk in bulk, as is done with silk worms. Science will have to find a way to make synthetic spider silk if we are to take advantage of its wonderful properties. The key to this is to look at what spider silk is made of: protein! Protein A protein is a molecule composed of polymers (a compound of several repeating units) of amino acids bonded together. A protein is like a blob of spaghetti, made up of a very long sticky spaghetti noodle consisting of a chain of amino acids. Depending on how these amino acids interact with each other and surrounding water molecules, the protein chain folds up into a three dimensional shape, which largely determines the properties of the protein. There are 23 different amino acids, and any number of these can be chained together in any order to form a protein molecule. Thus there are countless possible protein molecules. Protein chains found in nature range from just a few to many thousands of amino acids. Each one of these can be completely specified simply by writing down the order of the amino acids in the chain. Spider Silk Student 2 Scientists have developed a variety of technologies for synthesizing, or producing by artificial means, proteins. They continue to develop new and better techniques. For example, given a protein sequence that we would like to synthesize, it is possible to “program” microorganisms to synthesize these proteins for us. Scientists do this by building a DNA sequence that codes for the desired protein sequence. The ability to build this sequence is a technological achievement of no small note. They then insert the sequence into the genome of some bacteria such as E. coli (another major technological achievement) and allow the bacteria to build the protein. Because of significant laboratory research, we already know the amino acid sequences for many silk proteins. Additionally, research suggests that the technology to manufacture spider silk is not too far off. But perhaps we could do even better. What if we changed the amino acid sequence? Could we find a better sequence of amino acids that would yield even better silk, or make some other material with even more amazing properties? How can we determine a good amino acid sequence? One answer is to compare the amino sequences of different types of silk proteins and between different species of spiders. By doing this, it may be possible to identify patterns in the sequences that contribute toward specific properties of spider silk. We can then use this information to design better proteins! In this unit, we will study an algorithm called sequence alignment, which allows us to efficiently compare different sequences in a biologically meaningful way. This algorithm is one of the fundamental tools of bioinformatics—a field that has revolutionized biological research through the use of mathematics and computer science. While the main ideas of sequence alignment can be described in purely mathematical terms, getting the details right requires some understanding of molecular biology. Spider Silk Student 3 Unit Goals and Objectives Goal: Understand protein sequences and their role in identifying relationships among various species and organisms Objectives: Describe protein sequences in relation to DNA. Explain why the alignment of protein sequences is of importance to biologists. Understand the methods used by researchers to align protein sequences. Goal: Understand the use of lattices to represent the alignments of genetic sequences. Objectives: Represent a pair of sequences to be aligned as an appropriate labeled lattice. Interpret any path in that lattice as a particular alignment. Apply an algorithm to a labeled lattice to generate one or more optimal alignments of two given sequences. Goal: Use technology tools and resources to examine and analyze alignments of genetic sequences. Objectives: Be able to access and use the Biology Student Workbench (BSW) Internet resource to carry out alignments. Analyze output from the BSW program. Spider Silk Student 4 Lesson 1 Molecular Biology Essentials DNA Deoxyribonucleic acid (DNA) is the well-known double helical molecule that is the basis of heredity. DNA contains all of the information used in the development and functioning of all living organisms. The information in DNA is encoded using four different nucleotides: adenine (A), guanine (G), cytosine (C), and thymine (T). These nucleotides are connected together sequentially along a phosphate-sugar backbone. The order of these nucleotides in this sequence determines the information that can be used to manufacture ribonucleic acid RNA and protein molecules, which perform all of the functions of the organism. This information can be represented succinctly simply as a string of letters from the four letter alphabet A, G, C, T. The structure of a DNA molecule is like a spiral staircase, as illustrated in Figure 1.1. The molecule consists of two nucleotide strands. The phosphate-sugar backbones of the two strands form the sides of the spiral staircase. In the middle, each nucleotide from one strand is connected to a nucleotide from the other strand to form a base pair, which is analogous to a “step” of the spiral staircase. Notice that the figure shows only two types of base pairs: A-T, and C-G. This is because nucleotides come in complementary pairs: ‘G’ pairs only with ‘C’ and ‘A’ pairs only with ‘T’. Figure 1.1: Cartoon drawing of DNA. Illustration by Cornell University [Public domain], via Wikimedia Commons Because of this complementary pairing, the sequence of nucleotides on one strand is completely determined by the sequence on the other strand. In this sense, both strands contain exactly the same information. But only one of the strands is used to make RNA or protein. This strand is called the coding strand; the other is called the complementary strand. Spider Silk Student 5 The following figure shows the two strands of a DNA molecule, but some of the entries in the complementary strand are missing. Can you fill in the missing entries? coding strand: complementary strand: C G A T A T G C T A A T A T C G G C G C C G A _ A _ T _ G _ G _ C _ The complementary base pairing enables the DNA molecule to be replicated. During cell division, the two strands of the DNA molecule are separated. Each of these strands serves as a template from which a copy of the other strand is reconstructed by attaching the complementary nucleotide to each nucleotide in the template. This results in two copies of the original DNA molecule. Sometimes mistakes, or mutations, happen during DNA replication. There are three main types of mutations: substitutions, insertions, and deletions. These three types of mutations play a big role in how the sequence alignment algorithm works. A gene is a segment of the DNA molecule that contains the information needed to make a particular protein. In humans, genes vary in size from a few hundred nucleotides to more than 2 million nucleotides. There are many thousands of genes in each DNA molecule. It is estimated that humans have 20,000-25,000 genes. RNA The information encoded in a gene is used to construct a protein molecule. The first step is to copy the information from the DNA into a messenger RNA (mRNA) molecule through a process called transcription. Like DNA, RNA (ribonucleic acid) is composed of nucleotides. But there is one difference: RNA uses the nucleotide Uracil (U) instead of using Thymine. Thus, the alphabet for describing RNA molecules consists of the letters A, G, U and C. Each RNA nucleotide pairs with a complementary DNA nucleotide: ‘A’ pairs with ‘T’, ‘G’ pairs with ‘C’, ‘U’ pairs with ‘A’, and ‘C’ pairs with ‘G’. A complex of proteins called an RNA polymerase, which acts like a robot, performs the transcription process. Starting at the beginning of a gene sequence, the RNA Polymerase moves along the coding strand of the DNA attaching a complementary RNA nucleotide to a growing RNA strand (See Figure 1.2). This process is repeated over and over again, producing many mRNA molecules from a single copy of the DNA. Spider Silk Student 6 Figure 1.2: Transcription from DNA to RNA and translation from RNA to protein. Note in the figure that the RNA molecule is identical to one strand of the DNA (except for the U replacing T) but is the complement of the strand from which it is actually transcribed. In a complement, G and C are switched, and A and T are switched. The following sequences show a piece of the DNA coding strand for a gene, and the mRNA that is being transcribed. Fill in the missing nucleotides for the mRNA. DNA coding strand: mRNA: C G A U A U G C T A A U A U C G G C G C C G A _ A _ T _ G _ G _ C _ Proteins The information in the mRNA is next translated into a protein molecule. Recall that a protein molecule is a sequence of amino acids. There are 23 principal amino acids, so fortunately there are enough letters in the alphabet so that each amino acid can be encoded with a single letter. Translation is performed according to a genetic code. The units of this code are called codons, which consist of triplets of nucleotides. Figure 1.3 shows the 64 possible codons in a table format. Spider Silk Student 7 Second letter C U C A G Phenylalanine (F) UUA UUG Leucine (L) CUU CUC CUA CUG Leucine (L) AUU AUC AUA Isoleucine (I) AUG Methionine (M); initiation codon GUU GUC GUA GUG Valine (V) UCU UCC UCA UCG CCU CCC CCA CCG ACU ACC ACA ACG GCU GCC GCA GCG Serine (S) U Tyrosine (Y) UGU UGC Cysteine (C) C Stop codon UGA Stop codon A Stop codon UGG Tryptophan (W) G CAU CAC Histidine (H) Arginine (R) CAA CAG Glutamine (Q) CGU CGC CGA CGG AAU AAC Asparagine (N) AGU AGC Serine (S) AAA AAG Lysine (K) AGA AGG Arginine (R) GAU GAC Asparic acid (D) GAA GAG Glutamic acid (E) UAU UAC UAA UAG Proline (P) G U Alanine (A) A G Threonine (T) GGU GGC GGA GGG C U C Third letter First letter U UUU UUC A A G U Glycine (G) C A G Figure 1.3: The genetic code. To use the table, start with any DNA nucleotide triplet (for example CCT). Transcribe this to a mRNA codon by replacing all the Ts with Us (to get CCU). Read the triplet from left to right (first letter C, second letter C, third letter U) and follow along in the table. You should arrive at the box containing amino acid proline (P). Try another one. GAC leads to aspartic acid (D). Not all 64 codons specify a single amino acid. Additionally, there are three that do not specify an amino acid during translation. In humans, translation involves only 20 amino acids. Therefore, several mRNA codon triplets may result in the same amino acid. Note that three codons are called STOP codons. They are UAA, UAG, and UGA. Instead of translating to amino acids, they tell the ribosome, which is making the protein, to stop the translation. In other words, they indicate the end of the protein, which is usually before the end of the RNA. Similarly, there is another codon called the initiator or start codon. It is AUG and codes for methionine, abbreviated M. The start codon signals the ribosome to start the translation and causes the first amino acid in the protein to always be methionine. Spider Silk Student 8 ACTIVITY 1-1 Translating Triples Objective: Translate nucleotide codons to amino acids. Materials: Handout SS-H1: Translating Triples Worksheet 1. Below are the first 15 codons in a nucleotide sequence. Check to see that the first 10 codons are translated correctly, and then use the table in figure 1.3 to fill in the last five amino acids yourself. atg agt tgg aca gcg cga ctt gct ctt cta ttg ctc ttt gta gct M S W T A R L A L L 2. Translate the following codon sequences to amino acids. a. atg ccc tgt gga gcc aca ccc tag b. atg acg gag ctt cgg agc tag d. atg cgg ata aaa ata tcc aat tac agt c. atg agc cag tac acc aca atg 3. Why are there 3 nucleotides in a codon? Why not 2? Why not 4? 4. How would you translate a sequence of 20 or 40 codons? Describe a more efficient method for translation of long codon sequences. 5. Are there other ways to depict the genetic code? Find a different image or diagram and be prepared to share it with your class. From DNA to Protein To review, the coding process starts with DNA, which is transcribed into RNA, which is then translated into protein. Look back at Figure 1.2 to review this process. Actual photographs of the process are shown in Figures 1.4 and 1.5. In Figure 1.4, arrow “Begin” shows DNA strands while arrow “End” depicts RNA strands. The direction of transcription shows shorter strands to longer strands. As shown, the transcription can take place simultaneously at many places in a gene as multiple RNA polymerase molecules move in series along the DNA. Spider Silk Student 9 From Wikimedia Commons http://commons.wikimedia.org/wiki/File:Transcription_label_en.jpg Figure 1.4: Photomicrograph of RNA being transcribed from DNA. In Figure 1.5, the start of translation is at the upper right (arrow). Note how the protein strands are longer on the ribosomes that are further from the start of translation. As shown, many ribosomes can simultaneously move along the mRNA, resulting in many copies of the protein being produced at the same time. Note that translation similarly takes place simultaneously along the RNA, as multiple ribosomes move along the strand. Source: https://ib-biology2010-12.wikispaces.com/Transcription+and+Translation Figure 1.5: Ribosomes and growing protein molecules strung along an RNA strand. Spider Silk Student 10 Lesson 2 Structure and Function Structure One of the most challenging problems in computer science is to determine the 3-dimensional structure of a protein from its amino acid sequence. After the protein molecule is created, it folds up into a 3 dimensional structure that is determined by the attractive and repulsive forces between the amino acids and the water molecules in the cell. These forces are different between different amino acids, so changing the amino acid sequence changes the 3 dimensional structure of the protein. Thus, the shape of the protein is completely determined by the order of amino acids in the sequence. This problem of protein structure has captured the imagination of mathematicians and computer scientists for several decades, and is still not adequately solved. For example, using some of the world’s most powerful computers, it typically takes several weeks of computation time to predict the structure of a protein molecule consisting of only a few hundred amino acids. Unfortunately, the results aren’t always reliable! Because of this computational difficulty, it is unlikely that we will be able to design proteins from scratch. Instead we need to learn patterns from existing proteins. This is why it is important to be able to compare the sequences of different protein molecules. The places where the sequences are similar often correspond to similar features in the three dimensional structure. We will come back to this idea after we talk about sequence databases. Publicly available sequence databases are revolutionizing the study of molecular biology. These databases exist worldwide and are maintained by institutions with funding from the U.S. government and governments in other countries. For example, the U.S. National Institutes of Health (NIH) through its National Center for Biotechnology Information (NCBI) maintains the largest U.S. sequence database, called GenBank. Protein and DNA sequences discovered through government-funded research must, by agreement, be added to these databases. Once added, they are promptly shared worldwide. The description “publicly available” means just that. Anyone can look at the information and all that’s required is a computer with an Internet browser. Let’s take a look at some spider silk proteins in the GenBank database. ACTIVITY 2-1 Sequence Databases Objective: Use a sequence database to examine patterns of DNA Materials: Handout SS-H2: Sequence Databases Worksheet Computer (web site http://www.ncbi.nlm.nih.gov/[2]) Each sequence that is deposited in the GenBank database has a name given to it by the researchers and a special unique number called an accession number. We can use the name to look up the spider silk sequences we want. Later we can use the numbers to be sure we always refer to the same sequences. Two types of silk mentioned earlier are major ampullate, which has Spider Silk Student 11 the name MaSP1, and minor ampullate, which has the name MaSP2. We will use these names to find our sequences. 1. To go to GenBank, first google “NCBI” and click on the NCBI HomePage http://www.ncbi.nlm.nih.gov/.[2] Notice at the top of the page there are a pull down menu for which database to use, a blank space to enter your search item and a button labeled “Search.” In the pull down menu select “Nucleotide.” In the blank space enter “MaSP1 spider” (without the quotes) and click Search or press the Enter key. This will bring up a page showing the 21 MaSP1 or similar records of different organisms resulting from the search. The number “21” is used here, but it may not still be “21” by the time you perform this search. These databases are updated on a regular basis, by researchers worldwide. Click on the record labeled Accession: AM259067. This will bring up a page of information about the MaSP1 as shown in Figure 2.1. Figure 2.1: GenBank page from NCBI for MaSP1 in Euprosthenops australis.[2] Spider Silk Student 12 a. What is the name of the organism described on this page? b. The information was published in an article entitled “N-Terminal Nonrepetitive Domain Common to Dragline, Flagelliform, and Cylindriform Spider Silk Proteins.” N-terminal refers to one end of the silk protein and this article discusses common features of several different types of silk proteins. In what journal and year was the article published? c. Where is the protein sequence of amino acids? d. What does the entry labeled “CDS” stand for? e. What are the first twelve entries in the protein sequence and what do they stand for? f. What do you notice about the last four lines of the protein sequence? 2. Other lines under the “CDS” label give additional information. For example, the line “15..>1196” means that the protein sequence is derived from the portion of the DNA sequence starting at the 15th character and going beyond the 1196th character. The gene name, as we already know, is shown to be MaSP1 and the protein product is called “major ampullate spidroin 1 precursor.” a. What do you notice about the sequence shown in the section labeled ORIGIN? How is this sequence different from the protein sequence discussed above? b. What sequence is shown in the section labeled ORIGIN? c. How long is this sequence? 3. Sometimes researchers want just the sequences without all the additional information. These can be obtained by going to the top of the page, in the box labeled “Display,” and choosing FASTA. That is the name of a formatting that gives just the sequence, in this case, the nucleotide sequence, and a brief identification line preceded by the “>” symbol. “FASTA format” is often used as input to computer programs that compare sequences. Now return to the GenBank display. 4. Let’s confirm the translation of the nucleotide sequence for MaSP1. Look back at the printout. At the top of the CDS section (remember, coding sequence), is a note that says it runs from positions 15 to 1196. Look at the DNA sequence and find position 15. The sequence letters occur in groups of 10 in rows of 60. a. Starting at position 15, what is the first codon triplet? b. Convert this nucleotide to its mRNA nucleotide to find the start codon and then translate this codon to its associated amino acid. ______ ______ __________ DNA mRNA amino acid Spider Silk Student 13 Note the unique number which identifies this sequence in the database. It follows the word ACCESSION and is “AM259067.” The number is also repeated elsewhere. If you ever want to look this sequence up again, return to the NCBI HomePage. For “Search” choose Nucleotide. Then enter AM259067 in the “for” field click “Go” and follow the links. Practice 1. Use GenBank to find another sequence for MaSP1 from the same organism. Print out and label a copy of the page. Is the amino acid sequence the same? Describe. Sometimes only part of the sequence is shown. 2. Find another sequence for MaSP1 from a different organism. Print out and label a copy of the page. What organism did you choose? Describe how the amino acid sequence is the same or different as for Euprosthenops australis? 3. Find a sequence for MaSP2. Print out and label the page. 4. Get the FASTA format sequences for two versions of MaSP2. Print out and label the page. Comparing Two Sequences Let’s take a look at another MaSP1 sequence and see if it appears similar to the MaSP1 sequence of Euprosthenops australis. Figure 2.2 shows the MaSP1 sequence from the spider Latrodectus hesperus, otherwise known as the Western Black Widow spider. Western Black Widow Spider Photograph by B D (Flickr) [CC-BY-2.0 (http://creativecommons.org/licenses/by/2.0)], via Wikimedia Commons Spider Silk Student 14 Many people assume these two species are related by a common ancestral spider species living perhaps millions of years ago. Finding obvious similarity between the protein sequences would support this assumption. The degree of similarity could even tell us how long ago the common ancestor lived. Let’s look for that similarity. Figure 2.2: Page from GenBank NCBI for MaSP1 in Latrodectus hesperus.[2] Spider Silk Student 15 The protein sequence in Figure 2.2 is again listed under “CDS /translation”. Notice that both sequences begin with the amino acid methionine (M) as expected, but the next few letters are different. Below are the first 10 letters of the Euprosthenops australis sequence and the first 10 letters of the Latrodectus hesperus sequence. MSWTARLALL MYS LS IQSDF They don’t look similar at all. What should we do? ACTIVITY 2-2 A Tale of Two Spiders Objective: Finding similar amino acid sequences. Materials: Handout SS-H3: A Tale of Two Spiders Worksheet Computer 1. Search for the Latrodectus hesperus in the GenBank web site. 2. Look through the first 120 letters of the Euprosthenops australis sequence and the sequence to see if you can find any parts that look similar. Record what you find on a blank sheet of paper. In order to assist you in identifying similar sections, the two sequences are labeled S1 and S2 and every tenth letter is marked. Try to find portions of the sequence that match. Euprosthenops australis vs. Latrodectus hesperus S1 Euprosthenops australis 10 20 30 40 50 60 MSWTARLAL L LLFVACQGS S SLASHTTPWT NPGLAENFM N SFMQGLSSM P GFTASQLDD M 70 80 90 100 110 120 STIAQSMVQS IQSLAAQGRT SPNKLQALN M AFASSMAEIA ASEEGGGSLS TKTSSIASAM S2 Latrodectus hesperus 10 20 30 40 50 60 MYSLSIQSDF PTTTMTWSTR LALSFFAVIC TQSIYALGQG NTPWSTKANA DNFMNGFLSA 70 80 90 100 110 120 CAQSGVFSAD QVDDMTTIGK TLMIAMDKMG GKISSSKLQA LDMAFASSVA EIATAEGGAN Spider Silk Student 16 3. Look at the first 10 letters in sequence S1 and the ten letters starting at position 15 in sequence S2. S1: M S W T A R L A L L S2: M T W S T R L A L S a. Is this a good match? b. Place a star below the sequence letters that match. What do you notice about the stars? Since we started 15 places into sequence S2 to find matches, this suggests that S2 is longer on the left than S1. 4. Record and align the entries S1 : 84 – 100 with S2 : 97 – 113. Did you identify these two portions of the sequence in part 2 above? S1: S2: a. Is this a good match? b. Mark the matching amino acids with a star. What can you say about the similarities of these two species? The results of your matching further suggest that these particular parts of the sequences may be essential for the properties of the silk proteins because they have remained essentially unchanged over millions of years during which the spiders and their proteins have diverged. We say that these amino acids have been conserved. Alignment Obviously, looking through sequences to find similarities is tedious. We can use computers to do it quickly and without errors by a process called alignment. This will be the subject of the remainder of this unit. It is important to note that the success of an alignment program is dependent on an understanding of the types of mutations that typically occur as proteins evolve. We have seen the two most common types of mutations: • Substitution. In this case, one amino acid is replaced by another. In the first comparison above, there were 4 substitutions: S T, T S, A T, and L S. We might accurately conclude from this that S and T frequently participate in substitutions. In the second comparison, there were 2 substitutions: N D and M V. • Insertion/deletion. This occurs when a small piece is added or removed from one of the sequences. In the first comparison in S2, there was an insertion of the first 14 amino acids. That is why the matching started at amino acid 15. Spider Silk Student 17 A complete alignment of the sequences S1 and S2 is given in Figure 2.3. An alignment program called CLUSTALW (we will examine this program in a later lesson)[3]. The line under the sequences codes the alignment as: * (asterisk) indicates a match : (colon) indicates a common substitution . (period) indicates a less common substitution - (dash) indicates an insertion or deletion. Figure 2.3: Alignment between the MaSP1 sequences in two spider species. Alignment has proven to be a powerful tool in researching the causes of disease. An example in humans involves the hemoglobin gene. Hemoglobin is the protein that carries oxygen in red blood cells. Alignment of the hemoglobin gene from a healthy individual and from an individual with sickle cell anemia shows a single substitution in the nucleotide at position 17 in the gene. In a healthy individual, the 6th codon is “gag,” but in an individual with sickle cell anemia this codon reads “gtg.” This results in a replacement of the amino acid glutamic acid (E) in the healthy hemoglobin protein with valine (V) in the sickle cell protein, which ends up giving the protein its unhealthy properties. Spider Silk Student 18 Alignments with more than two sequences are possible and can give more information about conserved amino acids, that is, those amino acids that have not changed. The conserved portions of the protein sequences are believed to be the most essential for protein function since they have not mutated over the millions of years of evolution that separate the species. In the alignment in Figure 2.4 a sequence has been added from a third spider, Argiope trifasciata, the Banded Garden spider. Photograph by Thomas Quine (Garden-Spider-2 Uploaded by High Contrast) [CC-BY-2.0 (http://creativecommons.org/licenses/by/2.0)], via Wikimedia Commons Notice that not only is the species different, but this is the sequence from the minor ampullate silk protein, MaSP2. Note the very strong conservation in the ‘KLQALNMAFASSMAEIA’ region. This clearly indicates an important role for this part in these proteins. Figure 2.4: Alignment between partial MaSP1 sequences in two spider species and the MaSP2 sequence in a third. Spider Silk Student 19 Using Alignments to Predict Protein Structures One final use for protein alignments is in predicting three-dimensional protein structure. Understanding the structure is essential for understanding the properties of silk proteins. Determining structure is a long, costly laboratory process. The number of known structures is only in the thousands while the number of proteins is in the millions. The known structures are stored in the Protein Data Bank[4] or PDB found at http://www.rcsb.org/pdb/home/home.do.[5] Unfortunately, no one has yet worked out the structures for spider silk proteins. This is not unusual. In fact, it puts us in the position of scientists who study a new protein. We can get some information about structure by asking which proteins in the PDB are similar to the spider silk proteins. This question is answered by finding proteins that align well with all or part of the spider silk proteins. Searching the PDB yields several proteins that align with a small part of the Latrodectus hesperus MaSP1 protein. Figure 2.5 shows the alignment with one of those proteins, subtilisin Carlsberg, from the bacteria Bacillus licheniformis, abbreviated ‘1c3l’ (that’s one cee three el). The alignment in Figure 2.5 shows another commonly used format that differs from that in Figure 2.4. Letters in the middle of the alignment denote matches. Plus signs (+) indicate common substitutions. Figure 2.5: Alignment of part of MaSP1 from Latrodectus hesperus (top sequence) with subtilisin Carlsberg (bottom sequence). Figure 2.6 shows the three-dimensional structure of subtilisin. The spider silk protein may share some of the structural features of this protein. In this “ribbon” image, the corkscrew shapes are alpha helices and the arrows are beta sheets. These are common protein structures. Licensed under Public domain via Wikimedia Commons - http://commons.wikimedia.org/wiki/File:1st2.png#mediaviewer/File:1st2.png Figure 2.6: Three-dimensional structure of subtilisin. Spider Silk Student 20 Lesson 3 Dynamic Programming Have you ever been travelling to school or to go shopping and run into a detour? You have to adjust your route. Perhaps this new route is not any longer, just different than the one you normally travel. If you wanted to travel a different route every day, how many days would it be until you took the same route to school twice? ACTIVITY 3-1 The Path to Work Objective: Explore the pattern of determining minimum paths. Materials: Handout SS-H4: The Path to Work Worksheet Sally, a hard-working storekeeper in the city of Mandicy, has an unusually curious mind and wonders about things that you and I might not realize need wondering about. Recently, she discovered that there were 210 different ways that she could walk from her apartment to her store. Believe it or not, this discovery has some bearing on our interest in spider silk. Here’s a picture of Sally’s portion of the city of Mandicy. Her apartment building is located at “A,” her store is at “S.” Two friends from her apartment also have stores near hers: Ted’s store is at “T” and Rita’s store is at “R.” 1. How many blocks is the walk from Sally’s apartment to her store? Try a few different routes and decide if the number of blocks is always the same. 2. Is the number of blocks found the minimum? The maximum? Explain. 3. Estimate without doing any calculations or making any lists how many ways there are for Sally to walk to her store in the minimum number of blocks. Explain your reasoning. One day Sally made a list of all the ways that there were, but that got very tedious. At first she got “214" as the answer, but then she found that she had duplicates in her list. When she got rid of the duplicates she had 207 ways, but she wasn’t completely confident that she hadn’t missed any others. Finally, she settled on a list of 210 ways. She was confident about this number, but was sure there must be a simpler way to figure this out. Spider Silk Student 21 4. Describe how you think Sally finally calculated the number with which she was confident. Frustrated, Sally walked a block north to the store of her friend Ted and showed him her work. Right away he said that the answer had to be at least 84. Stunned, she asked how he could know such a thing. She discovered that she wasn’t the only curious person in her apartment building. It turns out that years earlier, Ted had gotten curious about the same question and discovered that there were 84 ways to walk from their apartment building to his store. He shared that he had made a very systematic list of all possible ways. In fact, Ted still had his list and handed it to Sally. Ted’s list, like the ones Sally had made, used “E” and “S” to denote walking a block east or a block south. 5. If there are 84 ways to walk from the apartment building to Ted’s store, why does that mean that there must be at least 84 ways to walk from the apartment to Sally’s store? 6. In comparing her list to Ted’s, how can Sally easily pick out her paths that should also be on Ted’s list? Sure enough, there were exactly 84 paths on Sally’s list that passed by Ted’s store. What about all the other routes? There must be 126 of them, since she was confident about the 210 routes on her list and 210 – 84 = 126. A portion of Sally’s list, with some of those passing Ted’s store circled, is shown in the figure below. Spider Silk Student 22 7. What do you notice about all the non-circled routes? Describe the paths that these routes represent. Sally wondered if there was a convenient way to check if 126 routes pass by Rita’s store. Could it be possible that Rita had also thought about this problem? Unbelievably, Rita had done that calculation, shortly after hearing Ted’s story about his counting experience. She was quick to point out that she was in a much more accessible location than Ted, for her list had a full 50% more paths on it than Ted’s. That’s just the response Sally needed. 8. Why was Sally so excited with Rita’s statement? Sally took Rita’s list and put it next to Ted’s. And there they were—all 210 paths that she had found: 84 from Ted’s list and 126 from Rita’s list. She simply added an “S” to the end of each of Ted’s and an “E” to the end of each of Rita’s, put them together and presto! Practice 1. Complete the following grids to get a feel for Sally’s adventures on a slightly smaller scale. For each figure, make list of all the ways to walk from the top-left corner to the bottom-right corner, taking eastward and southward steps only. You can encode your different ways to walk using the letters “S” and “E.” One of the paths is given for each figure. a. EEES b. EESS c. EEESS 2. Now match up each way to walk in the grids on the left with one of the ways to walk on the grid on the right, in the fashion that Sally did in our story. Counting Walks on a Grid The problem that Sally was trying to solve is an example of a problem that is easily solved by a method called dynamic programming. When using this method, we attempt to solve a given instance of a problem by showing how the solution to that problem can arise from solutions to similar, but smaller problems. For example, Sally saw that the number that she was looking for Spider Silk Student 23 as the solution to her problem, 210, was the sum of the solutions to two simpler problems, namely those that Rita and Ted had already solved. Their problems were similar to hers, but were both simpler because they involved counting paths to a location that was nine blocks away instead of ten. In fact, Sally could have gotten her answer without making a list at all, if only she had known that Ted and Rita had already solved the problems for their stores. She could have just called them, asked for their numbers and added them. You might then ask, how could Ted have gotten his answer? Who would he have called? Think about this for a moment. Look at the figure below and decide whom Ted could have called in order to be able to compute his answers without having to make his own list. Did you conclude that Ted could ask g and h for their numbers, and then add them? If so, you were correct: There are 56 ways to walk from A to g, and 28 ways to walk from A to h, and 56 + 28 = 84. Whom should Rita call if she wanted to compute her answer of 126 the easy way? So that you can check your answer, I’ll tell you that there are 70 ways to walk from A to f on that grid. Did you get the right answer? In general, then, we see that to obtain the number of ways to walk from A to any corner on this diagram, we simply need to obtain the answers at the corners north of and west of our target corner, and add them together. Does every question reduce to finding the answers to simpler questions? Let’s consider a corner at the top of our diagram, such as corner m. Someone owning a store on this corner would have a very easy time determining the number of walks from A to his or her store. Remember, we count only those walks that don’t “backtrack,” Spider Silk Student 24 that is, that make the trip in as few blocks as possible. For corner m, this would be six blocks and there is exactly one way to walk from A to m on that grid, without backtracking. A person making a list like Ted’s at corner m would have a list with one entry: EEEEEE. In fact, for every corner along the top of that diagram, there would only be one way of walking to that corner in the least blocks possible. The same would be true for the corners along the left side of that diagram. There is exactly one way to walk the fewest blocks to each of those corners from A as well. These corners are called the “base cases” of our dynamic programming solution. You could imagine Sally making two phone calls, to Ted and Rita, who say “hold on, I’ll call you back with the answer in a moment.” And Ted and Rita each make two phone calls, and so on. But when the call gets to a corner at the top of our diagram, such as vertex m, that person does not need to make any call. He can simply say “one.” And the same would go for each vertex along the left side of the diagram. Pretty soon, Ted and Rita will get responses (Rita, from f and g, Ted from g and h), compute their answers and call Sally back, and then Sally can finally compute the answer to her question. We are now in a position to be able to compute the answer to Sally’s question pretty quickly, by hand, without making lists of any sort. We will simulate this frenzy of phone calls on paper, but only those phone calls that actually give an answer. Thus we will not simulate Sally making a phone call until after Rita and Ted have already obtained their answers. This is another key characteristic of dynamic programming algorithms: Asking the questions in the right order, so that the answers to our questions have already been computed! We’ll assume that Sally has initiated a whole cascade of phone calls full of questions, and we’ll start filling in the answers as we can compute them. The first set of easy answers we can fill in are those along the top and along the left. To each of these corners there is exactly one way to walk from A, without backtracking. So let’s put “1" at all of those corners: That’s a pretty good start. We’ve been able to label eleven corners with their answers--only 24 corners to go. Among the remaining corners there is only one whose answer we can compute from the answers of its neighbors. Do you see which corner that is? Label this corner with its answer. Spider Silk Student 25 The corner one block south then one block east of A is the one we can label at this point. That corner would ask its neighbor to the north, “How many ways to your corner?” and get the answer “one.” He’d also ask his neighbor to the west and get the same answer. Then adding those answers he’d discover that there are 1 + 1 = 2 ways to walk from A to his own corner, and this is obviously the right answer, since “SE” and “ES” are the only two ways form A to that corner. This allows us to label one more corner: Now there are two corners who can query their neighbors to get their answers. Find them, and fill in the answers at those corners. Finish this grid by filling in the numbers at all the corners, including Sally’s corner. If you get 210 for her corner, then you know you’ve done all your arithmetic correctly. Practice A More General Situation. Suppose that Sally lived in a more interesting city, in which there might be any number of blocks leading into a given corner. Such a city is shown below. 3. Label the corners in this diagram with the total number of ways to walk to each corner. Spider Silk Student 26 4. List all of the ways to walk to the corner labeled with a square and all of the ways to walk to the corner labeled with a circle (one example is done for each corner). To Square: ESS, To Circle: EES, Shortest Paths After Sally had figured out how to do dynamic programming, she decided to try to solve a more useful problem: “What is the shortest route from her apartment building to her store?” By “shortest” Sally means “the least number of steps.” To help her answer this question, Sally walked every single block between her apartment and her store, counted the number of steps, labeled each block on her diagram and studied the result. Sally’s diagram is reproduced below. The diagram represents a significant amount of data. As we’ve already seen, there are 210 different ways for Sally to walk from her apartment to her store, so how should she determine the shortest path? Naturally, Sally would like to find a better method than “check them all” to find the shortest route. Questions for Discussion 1. Why are the blocks labeled with different numbers? Explain at least 2 reasons. 2. Brainstorm possible methods for Sally to find the shortest route to her store. 3. Will the shortest route to Sally’s store necessarily be one of the 210 routes with the fewest blocks? Explain why or why not? Spider Silk Student 27 The idea that eventually worked for Sally was very similar to the method she used for counting the paths. Her thinking went like this: Suppose I call Rita and Ted again, and see if they have solved this problem. That wouldn’t be quite as helpful as in the counting problem, because their steps are not the same length as mine. But suppose, just for supposing’s sake, that they knew exactly the least number of my steps that it would take for me to get to their stores, along the best possible path. What could I do with that information? Suppose the shortest path to Rita’s store takes 586 of Sally’s steps. You still do not know the actual shortest path, but you know the shortest path will take 586 steps. And suppose you find out that the shortest path to Ted’s store took 579 of Sally’s steps. Can you use that information to find the length of the shortest path to Sally’s store? Look over Sally’s Steps Map and figure out the least number of steps to get to Sally’s store. The reasoning goes as follows: To walk to her store, Sally has two choices regarding what her last block to the store ought to be. Either she walks from the north, via Ted’s store, or from the west, via Rita’s store. • The distance to Sally’s store from Ted’s store is 74 steps, and if we add that to 579 steps, which is the shortest possible way to get to Ted’s store, we get a total of 653 steps. • The distance to Sally’s store from Rita’s store is 65 steps, and if we add that to 586 steps, which is the shortest possible way to get to Rita’s store, we get a total of 651 steps. • Since 651 is less than 653, we ought to go via Rita’s store. We conclude that the shortest path to Sally’s store consists of 651 steps and passes by Rita’s store. This is another form of dynamic programming. Sally wanted to find the least number of steps to her store, and was able to do so by obtaining the solutions to two smaller problems and using them to find the solution to her problem. Of course, the question remains as to how Sally could have obtained the solutions to those two smaller problems. Imagine that Ted, Rita and all the other storekeepers took Sally-sized steps, and that they all had access to Sally’s Steps Map. Then each storekeeper could call their two neighbors, ask for the least number of Sally-sized steps to walk from A to those neighboring stores and then use that information to find the least number of steps from A to their own store. Once again, we have phone calls going in one direction (toward smaller, easier problems) and answers coming back in the other direction. Let’s see, for example, how we determined that the shortest path to Rita’s store is 586 steps. When Rita asked her neighbor to the west for the length of the shortest path to that store, she got the answer 519. Rita does not have to worry about how the shopkeeper obtained that answer. She just takes it as a given that if she comes from the west, the best she can do from that direction would be those 519 steps, plus the additional 67 steps along the block to her west, for a total of 586. Similarly, she asks the shopkeeper to the north for the shortest path to his store, and gets the answer 518. She then concludes that the shortest path to her store coming from the north would be 518 + 79 = 597 steps. But since this is greater than the number of steps she can obtain by coming from the west, that is, 586, Rita concludes that 586 is the best possible. Spider Silk Student 28 How did Ted obtain his number? The only piece of information you need that you don’t have yet is that the shortest path to the store on the corner north of Ted’s store is 538 Sally steps. So, just as we did with the counting problem, we can solve the “shortest path” problem at any given corner by having the shopkeeper at each corner ask his two neighbors for the answers at their corners, and use this information, together with the Sally’s Steps Map, to find the answer at our given corner. In the case of the counting problem, we had some corners that were “base cases,” that is, corners that did not need to place other calls to get their answers. What is the situation in our present problem? Consider the figure below, which shows the top row of Sally’s Steps map. If one were to ask the rightmost corner shopkeeper in that map for the length of the shortest path to that corner, she could not answer immediately. Although the only path to that corner is along the top row of the map the total number of steps needed to get to that rightmost corner depends on all of the numbers in that top row. The shopkeeper has two ways to determine the least number of steps. One way is to consult Sally’s Steps Map and add all the numbers in the top row. Another way is to simply do what all the other corners do: Call her neighbor, get the neighbor’s answer, and then add 64, which is the number of steps along the block to his west. Note that since hers has no block to her north in this diagram, she needs to call only one neighbor instead of two, and she does not need to select the minimum, since there is only one way into her corner. Shopkeepers along the north-most row can all do it this way, and the west-most shopkeepers determine the minimum steps to their stores by calling only their neighbor to the north. The numbers have to start somewhere. Which storekeeper(s) can definitely answer the question “What is the length of the shortest path to your store?” right away, without making any calls or doing any computation? Anyone calling the storekeeper at corner A where Sally’s apartment building is located would immediately receive the answer 0, since Sally needs to take no steps to get from her apartment building to her apartment building. So this corner is the only “base case” for the shortest path problem. We are now in a position to find the lengths of the shortest paths to all the corners in the Sally’s Steps Map. The numbers for a few blocks in the northwest corner of the map are shown. Spider Silk Student 29 In the figure above, the bold numbers at each corner show the length of the shortest path to that corner, while the numbers along the blocks indicate the number of steps Sally must take to traverse that block. Additionally, the thickened path indicates those streets along which the shortest route to a particular corner travels. For example, the path EESS results in the shortest path of 264 steps to that resulting corner. The shortest path came from the north, leaving the road from the west unshaded. Practice 5. Complete Sally’s Steps Map and determine the actual shortest path from her apartment to her shop. Extension 1. A More General Situation. Suppose that Sally lived in a more interesting city, in which there might be any number of blocks leading into a given corner as shown below. This city diagram includes a few of the shortest path numbers for you and shades those blocks giving the shortest route into a corner. Note that to find the corner labeled 22, we had to compare three different sums from three different corners, and select the minimum. Also, when computing the number 15, we had a tie score. What do you notice about the blocks into that corner? Complete the diagram to find the shortest path from A to S. List the directions associated with the shortest path and the number of steps. Spider Silk Student 30 2. How many ways are there to walk from A to B on each of the figures shown below, where each step must move either east, south or diagonally southeast? A A B B Figure 1 Figure 2 A A B B Figure 3 Spider Silk Figure 4 Student 31 3. The numbers on the edges of the graph below represent distances. What is the length of the shortest path from A to B? How many routes achieve that length? 4. What is the length of the shortest path from A to B on each of the figures below, where each step must bring you closer to B? Spider Silk Student 32 5. Find the length of the shortest path from A to S on the map below, and also identify the shortest path by shading the appropriate edges. This problem is pretty close to ultimate problem we will be addressing regarding spider silk! Spider Silk Student 33 Lesson 4 String Alignment You might be wondering how the previous lesson’s topics of paths and shortest distances relate to spiders and DNA. You will soon see that you are in a good position to solve some DNA problems. Mutations Recall the basic theory of DNA evolution: A long time ago in a species far, far away there was a gene X that described some protein that contributed to the life and health of that species. Over the millennia this gene, X, together with the thousands of other genes in this species, changed in various ways, leading to several different species that we can see today, all of which are descended from that one ancestral species, and all of which carry some remnant of the gene X. The diagram above gives an example (on a very small scale) of this type of action. At the top of the tree diagram is the gene X in some now-extinct, ancestral species a long, long time ago. The two genes just below that in the tree represent how the gene X looked in two species, also now extinct, a long time ago. At the bottom of the tree we find how the gene X looks in four species that are alive today. The variations seen among these genes today have arisen by various mutations through the ages, as organisms passed their genetic material (DNA) to their offspring. These mutations consist mainly of insertions, deletions and substitutions, as we discussed in Lesson 1. Let’s take a look at how the sequence at the top of the tree diagram mutated to become the sequence below it, on the left. Here, the sequence “AATTGGGGCCCCA” became “AATTGGGCCTCA.” One likely explanation is that one of the G’s got deleted, and that the third C mutated to a T. The diagram below represents this explanation. Spider Silk Student 34 In this representation, the various nucleotides are aligned in columns to show which nucleotide in the child species corresponds to each nucleotide in the parent species. We can see how the G was deleted, because in the child species (below) there is a dash where the parent has a G. And beneath one of the C’s in the parent, there is a T in the child. The alignment diagram below shows one possible relationship between the sequence at the top of the tree and its child on the right. In this diagram, the hyphen “-“ in the top string represents a gap. This gap could be due either to a deletion in the 1st string, or to an insertion in the 2nd string. In this example, we know it was an insertion in the 2nd string because we know that the 2nd string evolved from the 1st string. But if we don’t know the history, there is no way to know, just from comparing the strings, whether the difference is caused by insertion in one string or deletion in the other. So, we use the gap symbol to represent either of these possibilities. In this case we also see one substitution mutation, where a T in the parent became an A in the child. And we see one insertion mutation, where a G was inserted into the sequence in the child just before the last A in the gene. Now, you might have noticed that these are not the only possible ways to explain the observed mutations. The following diagram shows three additional alignments that might explain the relationship between the sequence at the top of the tree and its left child. All three of these explanations are theoretically possible, though they seem to become progressively less likely as you move from left to right across the table. Option 1 is essentially the same as that given in the previous discussion, but the first G was deleted here while the fourth G was deleted above. Option 2 also has a single deletion, but uses two substitutions to explain the changes. Option 3 has many changes occurring. Spider Silk Student 35 Questions for Discussion 1. Are there other options that could explain the mutation from the top parent species to the second level left child species? If so, list two. 2. Why is it that Option 1 feels more “right” than Option 3, even though, as far as we know, either one could be historically accurate? The generally accepted principle of Occam’s Razor asserts that, all other things being equal, people prefer simpler explanations. For biologists, this principle, usually call the parsimony principle, is heavily relied upon to make our best guesses about historical events that we have no way of discovering exactly at this time. But gut “feelings” are hard to turn into computer programs. What is needed is a scoring system. A scoring system is an objective way to attach a numerical value to the quality of an alignment. A sample scoring system to score the first four alignments given above of the sequence of the top of the tree and its left child is as follows. In each column of the alignment score: +2 for each match –1 for each mismatch –2 for each gap. For example, consider the alignment of the top sequence and its child to the right: AATTGGGGCCCC-A AATAGGGGCCCCGA To score this alignment, we consider each column of the alignment separately and assign a value to that column. The first three columns are matches, so each receives a score of +2, according to the scoring system we gave. The fourth column, however, has a T in the first string and an A in the second string, which is a mismatch, meaning that a mutation happened. Our scoring system assigns this a value of –1, since we want our alignments to prefer matches over mismatches. This reflects that successful mutations are rare. All of the scores have been entered in the table below. Note the penultimate column, where we have put a gap into the first string to show an insertion mutation. This column has a score of –2 according to our scoring system because insertions are even rarer than substitutions. Spider Silk Student 36 Adding up the scores for each column we obtain a total score of 21 for this alignment. Score each of the three alignments of the top sequence to its child below to verify the total scores shown in the table. This scoring scheme gives us an objective way to say which alignments are better and which are worse. In this case, we would say that alignments that had a score of 19 were the best, while the others were worse. But is 19 the best we can do? At this point, we do not know. Finding the best alignment is the subject of the next section. Alignments and Walks Now is when all of the hard work of the previous sections pays off. All that we have to do at this point is realize that string alignment is really a shortest path problem, and we’re done! Let’s find the optimal alignment of the strings “ACC” and “GGC,” where ACC is the initial sequence on top and GGC is the resulting sequence on the bottom. At the same time, let’s consider the table shown below. Spider Silk Student 37 Imagine that you are standing in the shaded square in this table as you are considering how to begin aligning these two strings. There are three possible ways to begin the alignment: Your first column can be either Suppose you chose , or . What do these options mean in the table? for the first column of your alignment. This would correspond to taking an east step in your table, because you would have “used up” the A in the top string, but used none of the letters of the second string. And if you chose for the first column, that would correspond to taking a south step, because you would not have used any letters from the top string, but would have used the G from the bottom string. Choosing for the first column would correspond to taking a southeast diagonal step, because that column used up the first letter of both strings. Suppose that we selected as the first step of our alignment, stepping east in our grid. Now we must select the second column of our alignment. Again, we have three choices: , Suppose we use or , corresponding respectively to an east, south or diagonal step. for this column, stepping diagonally in our grid. Continuing in this fashion, we might arrive at the alignment , for which the path is shown below by the shaded squares. Please take a moment to make sure that you see how the Spider Silk Student 38 alignment above and the walk shown below correspond with one another. Is there a better path than the one shown above? Each of the choices in the alignment has an associated score. Can you explain how you would obtain a score of -3? Starting at the upper left box again, you get –1 if you use column mismatch, and –2 for either of the choices or , because that is a , because they use gaps. We can include this information in the original table by writing the scores of taking each type of step in the space between the corresponding squares. Thus in the table below, on the left, we see that to walk east or south from the initially shaded square we incur a score of –2, while walking on a southeast diagonal from that square we incur a score of –1. The complete table-full of scores is shown in the table to the right. Spider Silk Student 39 Questions for Discussion 3. Note that all the horizontal and vertical steps score –2. Why? 4. The diagonal steps are either +2 or –1. Explain why. 5. Find the score of the following alignment explicitly. Each column of the alignment corresponds to some step on our grid. a. corresponds to an East step, since we use only an A from the top string. What score does this step incur? b. corresponds to a Diagonal step, since we use a character from each string. What score does this step incur? c. corresponds to a South step, since we use only a character from the bottom string. What score does this step incur? Spider Silk Student 40 d. corresponds to another Diagonal step, since we use characters from both strings. What score does this step incur? 6. Using a copy of the scoring table above, shade the squares that we walk through in this alignment. Put into each shaded square the running total of the score of our alignment along the path. Note that this is similar to what we did in trying to find the shortest path to Sally’s store! ACTIVITY 4-1 A Corresponding Walk Objective: Understand the correspondence between string alignments and walking on the alignment table. Materials: Handout SS-H9: A Corresponding Walk Worksheet Shown here is another example of an alignment and its corresponding walk. Figure 4.1: Walk Lattice 1. Have one group member explain how the first column in the alignment corresponds to the walk shown above. Remaining group members take turns to explain each subsequent column and step in the walk. 2. Each of the choices made in the walk above has an associated score. Use the same scoring of +2 for each match, -1 for each mismatch and -2 for each gap. Write the running total of the score in the shaded boxes. What is the final score of the alignment walk depicted above? Spider Silk Student 41 Before finishing this section, make sure you understand the correspondence between string alignments and “walking” on an alignment table. Look back at Figure 4.1. Take note of the rows and columns: There is one extra row and one extra column before beginning the characters of the strings. This is required in order to give us a starting point for our walk, prior to using up any of the characters in the alignment. Also, every alignment of these two strings corresponds to some walk in the table from the northwest corner to the southeast corner, taking only east, south or southeast diagonal steps. And in the other direction, every such way to walk corresponds to some alignment. Thus, finding the optimal alignment of these two strings amounts to finding the optimal path. Practice 1. Give the alignment that corresponds to the walk shown in the table to the right. 2. In each of the shaded boxes, put the running total of the score of the alignment. Assume that we now award +1 for a match, –1 for a mismatch and –2 for a gap. 3. Find an alignment of these two strings that achieves a better score. 4. Show the walk corresponding to your improved alignment, by shading the walk in the table to the right. Spider Silk Student 42 The Optimal Alignment Algorithm You are now in a position to answer the question: “How can I find the optimal alignment between two strings?” Consider the same two strings examined previously and shown in the figures below. On the right you see the table that we have been using, and on the left you see a map similar to the walking maps from Lesson 3. Finding the alignment with the highest score is like finding the longest path from Sally’s apartment to her store. Finding the longest path to Sally’s store (without backtracking) is solved the same way as finding the shortest path, except that at each corner we select the greatest sum instead of the least sum. Don’t worry about the negative numbers on the “streets” of this map. Even though a negative number has no physical interpretation in terms of steps, it works fine for our mathematical computations of addition and then comparison to select the greatest number. The cumulative scores for paths along the top row, the left column and the first diagonal square are completed. Using the given scores for the first step, the greatest is -1 and so the optimal alignment would use this path. ACTIVITY 4-2 The Optimal Alignment Objective: Find the optimal alignment for two sequences Materials: Handout SS–H10: The Optimal Alignment Worksheet 1. Use the map below to find the highest scoring path from A to S. Note that we still do not allow backtracking, so that all travel must go in an east, south or southeast diagonal direction. Spider Silk Student 43 2. Repeat the activity above, except this time perform the computation directly on the table below. Fill in the numbers in the blank cells, using the numbers between the cells to tell the score from cell to cell. When you do string alignments in real life, you are not going to want to write in all of those scores between the cells of your table. There is no need to do so since we know that any east or south step will score –2, and Diagonal steps score either +2 or –1, depending on whether the letters in the row and column into which we are about to step match or mismatch. We have done this in the table below for the strings “AGCGT” and “CAGT.” Note that in addition to putting the numbers into each cell, we have also marked where the greatest value came from by putting lines between the cells, showing which “roads” we walked along to obtain the greatest value. To build an optimal string alignment from a completed table, we start at the southeast corner and walk back along our marked connectors until we reach the northwest corner. Note that trying to walk the other way can get us stuck at a dead end, without reaching the southeast corner. Spider Silk Student 44 Continuing the above example we find a way to walk back from the southeast corner to the northwest corner. This path is shaded and yields the string alignment: - AGCGT CAG - - T There are several ways to walk back along an optimal path. In this case there are six optimal paths altogether, including the one we found above. While they each give a different alignment, they will all have the same score, 0. Practice 5. What alignment is implied by the following scoring matrix? Spider Silk Student 45 6. How many optimal alignments are indicated by the following scoring matrix? 7. Here is the start of an alignment between "ACC" and "CGAA" with match score +2, mismatch penalty –1, and gap penalty –2. Complete the matrix. C G A A 0 | -2 | -4 | -6 | -8 — \ \ \ A -2 — \ -1 | \ -3 \ C -4 — \ 0 — | \ C -6 -2 –5 8. Give all optimal alignments between "ACC" and "CGAA" with match score +2, mismatch penalty –1, and gap penalty –2, using your work from the previous problem. Spider Silk Student 46 9. Find the optimal alignment of the strings AGT and TGA, using match score +2, mismatch score –7, and gap score –2. 10. Two DNA sequences derived from a common ancestor in an environment in which insertions and deletions were much more likely than point mutations. To reflect this in an alignment, a researcher assigns a match score of +3, a mismatch score of –1, and a gap "penalty" of +1. Here is the resulting scoring matrix. Complete the matrix. 11. Can you see why under the (very artificial) scoring system given in the previous problem, an optimal alignment of any two strings will never align two mismatched bases? In fact, what relationship between the gap penalty and the mismatch penalty will guarantee this behavior? Spider Silk Student 47 12. Consider the two alignments shown below of the two strings ACCGG and TATGACCGGTTGTG: The alignment on the left is preferable to the alignment on the right, because it preserves the integrity of our first string much better than the alignment on the right does, but our scoring system will give them equal scores. If we modify our scoring system so that it does not charge for gaps at the beginning or end, then the alignment on the left will have a much higher score, and will be preferred to the other alignment. This exercise will show us how to modify our algorithm accordingly. There are two strings: "AACCTT" and "ACTACT" a. Align them using the following scoring system: match = +2, mismatch = –1, initial gaps and end gaps = 0, and all other gaps = –2. The first few entries have been filled in for you, as has the final score, so that you can check your work. b. How many optimal alignments are there? c. Show the optimal alignments. Spider Silk Student 48 Lesson 5 Aligning with Biology Workbench The Student Interface to the Biology Workbench (SIB)[3] is a Web-based bioinformatics resource. It provides a set of powerful tools to investigate problems in molecular biology—the same tools used by research scientists. In the first activity of this lesson you will look at proteins that make up the silk of two species of spiders. In the second activity you will add three more proteins from three other species of spiders to your analysis. ACTIVITY 5-1 Introduction to Using Biology Workbench Objective: Introduce you to the Biology Student Workbench. Materials: Handout SS-H11: Introduction to Using Biology Workbench Worksheet Computer 1. Go to the Student Interface to the Biology Workbench (SIB) website at http://bighorn.animal.uiuc.edu/cgi-bin/sib.py[3]. a. Set up an account by following the instructions to register on the screen. Complete your registration by supplying a user name and a password. b. Return to the SIB page and log in. Click on NEW (see 1st arrow in Figure 5.1) to create a new session. Name this session Spider Silk. Figure 5.1: SIB Page Shot 1 2. Scroll down to the bottom of the page and place a check (click) in the box to the left of the session that you just created. a. Scroll back up to the top of the page and click the button labeled PROTEIN TOOLS (see 2nd arrow in Figure 5.1). b. In the table on the protein tools page look for a row with a tool (button) called Ndjinn (see 3rd arrow in Figure 5.2). Spider Silk Student 49 c. You are going to search for a specific protein. In the cell to the left of this tool there is a search window (see 1st arrow in Figure 5.2). Type in Araneus gemmoides 1 tubuliform spidroin. Figure 5.2: SIB Page Shot 2 d. Next select a database to search by clicking (highlighting) GenBank Invertebrate Sequences (see 2nd arrow in Figure 5.2). e. Then click on the button labeled Ndjinn (see 3rd arrow in Figure 5.2). You should now have a search results screen that resembles Figure 5.3. f. Place a check in the box to the left of the match that has a rank of 0 (see 1st arrow in Figure 5.3). g. Check that the protein description matches what you typed into the search window note that it has a rank of zero. Figure 5.3: SIB Page Shot 3 h. Scroll down to the bottom of the page and click on Import Sequence(s) (see 2nd arrow Figure 5.3). You should now be back on the Protein Tools page (see Figure 5.2). i. Repeat these steps to search for and import the protein Nephila clavipes tubuliform spidroin. 3. Scroll down to the bottom of the page and select (click in the boxes) the 2 protein sequences that you just imported (See 1st arrow in Figure 5.4). Figure 5.4: SIB Page Shot 4 Spider Silk Student 50 To find out more about the animals these proteins come from click VIEW RECORD in the right hand column (not shown in Figure 4). Use the information on this page to answer the following questions about these animals. a. What is the tubuliform silk used for in both species of spider? b. Who was the researcher(s) who posted the amino acid sequence for both types of spider? c. Where were these researchers working when they submitted this information to this web site? d. What type of molecule was translated to produce the amino acid sequence in this protein? e. Are the molecule type, gene name, and protein name the same for both species? 4. Now click RETURN at the top of the page to go back to the Protein Tools page. You will now compare the two proteins you have selected. Scroll down to the bottom of the page and make sure both protein sequences are selected. Then click on the button labeled CLUSTALW (see 1st arrow in Figure 5.5). Figure 5.5: Protein Tools Page This page shows a comparison of the sequence of amino acids from the two species. Answer the following questions from this page. a. What do the blue letters mean? b. What do the asterisk, colons, and periods at the bottom of the alignment mean? c. How many amino acids are there in each protein? d. What is the alignment score? e. What scoring matrix was used in this alignment? 5. Before exiting this screen click on the button IMPORT ALIGNMENT, which should take you to a new screen. This is the screen showing the alignment tools available in Biology Student Workbench. Scroll down the screen and select CLUSTALW. Spider Silk Student 51 6. Click the button labeled BOXSHADE (see 1st arrow in Figure 5.6). Figure 5.6: Alignment Tool This display is a color-coded view of the alignment from the previous page. Answer the following questions about this page. a. What does the blue color mean? b. What does the green color mean? c. What does the yellow color mean? d. What does consensus mean? Amino Acid Scoring Matrices When we aligned our DNA sequences, we used a simple scoring system that had one score for matches, one for mismatches and one for gaps. When aligning amino acid sequences, a more interesting scoring system, such as the Gonnet matrix shown below is used. Figure 5.7: The Gonnet scoring matrix. The numbers in the scoring matrix are related to the probabilities of a particular substitution occurring and surviving in nature. Recall that the amino acid sequence of the protein is derived from the nucleotide sequence in the DNA. Thus, a change in the amino acid sequence is actually caused by a mutation in the DNA. Take a look again at Figure 1.3 in Lesson 1. Notice that codons for some amino acids differ by only a single nucleotide. For example, the codons for Spider Silk Student 52 Serine (S) (AGU and AGC) differ only in the middle nucleotide from codons for Threonine (T) (ACU and ACC). In contrast, the codon for Tryptophan (W) (UGG) has nothing in common with any of the codons for Asparic Acid (D). Thus, replacing a Serine with a Threonine occurs with higher probability than replacing a Tryptophan by an Asparic Acid. A much more important consideration, however, is whether the substitution actually survives in nature. Recall that changing an amino acid can cause a change in the three dimensional structure of a protein. If this change is large, it can completely change the properties of the protein. Most mutations are bad! If the protein performs an important function in the organism, the modified protein is no longer able to perform the work that it is supposed to do. As a result, the organism is less likely to mature and reproduce. Thus, while many mutations happen in nature, most of them don’t survive in the gene pool. It turns out that many pairs of amino acids have very similar properties; so substituting between them does not dramatically alter the protein. Thus, when we compare sequences found in nature, we are more likely to see substitutions between similar amino acids, and less likely to see substitutions between amino acids that have very different properties. The numbers in Figure 5.7 provide scores that take these considerations into account. A negative score means that the substitution is rarely found in nature, and a positive score means that it is relatively common. To score an alignment, we look up the two amino acids in the table to find what score to give if those two amino acids are aligned with each other. Then, as before, the score of an alignment is the sum of the individual scores. For example, the score for the alignment shown here would be 4.9, using a gap value of –5. (This gap value is independent of the matrix, and was an arbitrary choice for this example.) A A 2.4 M C –0.9 I I 4.0 N D 2.2 E –5 S S 2.2 Similarly, when using our dynamic programming alignment algorithm to align amino acid sequences, we would also use the scores from the matrix shown for any diagonal move (corresponding to aligning a pair of amino acids). The problem is no more complicated, but it is more tedious because we have to look up scores from a table. Computers, of course, do not mind this. Spider Silk Student 53 ACTIVITY 5-2 Comparing Spider Silk Protein Objective: To compare the amino acid sequence of the silk protein from five species of spiders. Materials: Handout SS-H12: Comparing Spider Silk Protein Worksheet Computer 1. Open the Student Interface to the Biology Workbench: http://bighorn.animal.uiuc.edu/cgibin/sib.py[3]. Login and open the session called “Spider Silk” that you created in Activity 5-1 by following the directions in the RESUME row (see 1st arrow in Figure 5.8). Figure 5.8: SIB Page Resume 2. Click on the button that says Protein Tools at the top of the screen (see 2nd arrow in Figure 5.8). The screen should now resemble Figure 5.9. Figure 5.9: SIB Page Protein Tools a. Type Uloborus tubuliform into the box that says “Enter your search in the box below” (See 1st arrow in Figure 5.9). b. In the box labeled “Ndjinn” select the following database: Genbank Invertebrate Sequences (see 2nd arrow in Figure 5.9). c. Click on the button labeled “Ndjinn” (See 3rd arrow in Figure 5.9). Spider Silk Student 54 3. Place this sequence into your session labeled Spider Silk by placing a check (click) in the box to the left of the protein selected (See 1st arrow in Figure 5.10). Go to the bottom of the page and click on Import Sequences(s) (See 2nd arrow in Figure 5.10). Figure 5.10: SIB Page Selection to Import 4. Repeat this procedure for Argiope aurantia tubuliform. This will return several sequences. Import the one with accession number 61387230. 5. Repeat this procedure for Deinopis tubuliform, importing sequence number 63054332. 6. Take a look at the five sequences at the bottom of your Biology Student Workbench page, and make sure they are the ones shown in Figure 5.11. To compare the five sequences that are now in your Spider Silk session, at the bottom of the page check all five sequences by placing a check (click) next to each. Figure 5.11: SIB Comparing Sequences 7. Now click on CLUSTALW in the tool column (See 1st arrow in Figure 5.12). This performs a multiple sequence alignment of the five spider silk proteins that we have imported. Note that we did not discuss how to do alignments of more than two sequences, but the basic idea is the same: Use dynamic programming. Notice that the first step in aligning these five sequences was performing all pairs of pairwise alignments. Figure 5.12: CLUSTALW Answer the following questions about the display on this page. a. What is the length of each of the five protein sequences, in order from longest to shortest? b. Find a stretch of five amino acids that is the same in all of the silk protein sequences, and aligned. What are they? Spider Silk Student 55 c. Which pair has the greatest pairwise alignment score? Write out the protein ID numbers of the two that have this greatest alignment score. d. Where is the output concerning the pairwise alignments? e. What is the overall multiple sequence alignment score? 8. Click on the “Import Alignments” button at the top of this page. (See 1st arrow in Figure 5.13.) This saves the work that CLUSTALW just did, so that we won’t have to perform this alignment again. We’ll come back here later. Figure 5.13: SIB Import Alignment 9. Click on the “Protein Tools” button at the top of the page. You should now be on the Protein Tools page. Scroll to the bottom of this page—if the five spider silk proteins that you previously imported are not still checked—check them (see Figure 5.11). Scroll up the page and click on the button labeled AASTATS (see 1st arrow in Figure 5.14). Figure 5.14: SIB AASTATS Answer the questions below. a. For each of the five spider species list the two amino acids that appear the most frequently and how many times that amino acid appears in the spider silk protein. Protein Most common amino acid Name Freq. Percent 2nd most common amino acid Name Freq. Percent b. According to the list you generated in Question a, which two proteins are most alike? Spider Silk Student 56 c. According to the list generated in Question a, which two proteins are least alike? d. Which amino acids never appear in any of these sequences? 10. Click on the “Return” button at the top of the page, returning you to the “Protein Tools” page. At the top of the page, click the “Alignment Tools” button, bringing you back to the page where we saved our CLUSTALW alignment. At the bottom of this page, select the check box next to the names of our five spider silk proteins. Notice that there is only one check box there. This is because the alignment of those five sequences is now to be treated as one large object containing those five sequences as well as information on how they are aligned. 11. Click on the “DRAWTREE” button in the tools section of this page. This generates a graphic showing the relationship between our five sequences. In figures such as these, the lengths of the segments are used to indicate how different sequences are from each other. Thus, since the labels ending in “231" and “237,” corresponding to Argiope aurantia and Araneus gemmoides respectively, are the closest together on this tree, we conclude that their sequences are the most closely related. Indeed, these two were the pair with the highest pairwise alignment score, as you discovered in Question 3. Such trees are considered to be good guesses at the evolutionary relationship between the proteins, and perhaps the species they came from. Spider Silk Student 57 Glossary Adenine - one of four nucleotide bases, abbreviated as the letter “A” in a DNA sequence. Alanine - an amino acid found in spider silk, abbreviated as the letter “A” in a protein sequence. Alignment - the process whereby different DNA or protein sequences are compared. The sequences may be from the same or different individuals or from different species. Arachnida - the class where spiders are classified in the animal kingdom. This class includes scorpions, mites, and ticks. Araneae - the order where spiders are classified in the animal kingdom. This order contains thousands of spider species. Arthropoda - the phylum where spiders are classified in the animal kingdom. This phylum includes insects, arachnids, and crustaceans. Base case - a sub-problem whose solution is obvious by inspection. Codon - three letters from the nucleotide sequence that “codes” for an amino acid. Conserved - term given to nucleotides or amino acids in a sequence that have not changed over a long evolutionary time. Cytosine - one of four nucleotide bases, abbreviated as the letter “C” in a DNA sequence. Deletion mutation - the removal of a nucleotide from an ancestor’s DNA sequence. DNA molecule - a chain of nucleotides, usually double stranded. DNA nucleotide sequence - a chain of nucleotide bases – adenine (A), guanine (G), cytosine (C), and thymine (T). DNA sequence - see “DNA nucleotide sequence” Dynamic programming - a process for finding the optimal solution to a problem by systematically identifying and solving a sequence of similar sub-problems. GenBank - one example of a sequence database. Genome - all the DNA is an organism’s cell, usually all the chromosomes. Glycine - an amino acid found in spider silk, abbreviated as the letter “G” in a protein sequence. Guanine - one of four nucleotide bases, abbreviated as the letter “G” in a DNA sequence. Spider Silk Student 58 Insertion mutation - the addition of a nucleotide into an ancestor’s DNA sequence. Mutation - any change in a DNA sequence. Optimal - best according to some specific criteria. Parsimony principle - the principle that simpler explanations are more likely to be correct than are more complicated explanations. Protein molecule - a chain of amino acids. Protein sequence - a sequence of letters selected from the twenty-letter amino acid alphabet used to represent a protein molecule. RNA molecule - a chain of nucleotides, usually single stranded. RNA nucleotide sequence - a chain of nucleotide bases – adenine (A), guanine (G), cytosine (C), and uracil (U). Sequence database - a repository that stores the sequence information discovered in individual laboratories. Sequencing - the process by which a scientist extracts DNA or protein molecules from a subject and then treats the extraction in a laboratory to reveal the types and order of nucleotides (DNA) or amino acids (protein). Spider silk proteins - contain a high percentage of two amino acids, alanine and glycine. Substitution mutation - the replacement of one nucleotide of an ancestor’s genetic sequence with a different nucleotide. Transcription - the cellular process, involving RNA polymerase, which copies part of a DNA sequence into an RNA sequence. Translation - the cellular process, involving ribosomes, which converts the genetic code along an RNA molecule into a protein sequence. Spider Silk Student 59 References [1] John Wiley & Sons, Inc.. (2007, April 6). Fascinating spider silk. ScienceDaily. Found at www.sciencedaily.com/releases/2007/04/070405094039.htm. [2] National Center for Biotechnology Information (NCBI). Found at http://www.ncbi.nlm.nih.gov/. [3] San Diego Supercomputer Center. Biology WorkBench. Found at http://workbench.sdsc.edu. [4] Berman, H.M. et al. (1999). The protein data bank. Nucleic Acids Research. 28(1), 235-242. [5] RCSB Protein Data Bank. An information portal to biological macromolecular structures. Found at www.rcsb.org/pdb/explore.do?stuctureId=1c3l. Spider Silk Student 60