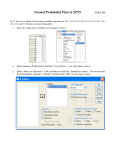
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Gene desert wikipedia , lookup
Genome evolution wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Genome (book) wikipedia , lookup
Genomic imprinting wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Microevolution wikipedia , lookup
Designer baby wikipedia , lookup
Gene expression programming wikipedia , lookup
Commonly Asked Questions: What are pre-processing and normalization and why do I need to do it? Raw microarray data extracted from image analysis contains many data points that are very low in overall intensity, saturated in both channels, noisy or otherwise are of poor-quality and questionable. Based on user-defined baseline and threshold values, such poor-quality features will be filtered out and removed from further processing and analysis which includes normalization, replicate averaging, etc. Normalization is used to adjust and balance individual signal intensities to reduce technical or systematic differences not caused by the treatment and allows for more meaningful interpretations of the biological effect of the treatment. Normalization is used to correct for systematic variation NOT biological variation. Systematic or technical variations include differences in: dye incorporation rates, RNA loading, RNA purity and quality, differences in laser age and power, emission characteristics and stability of the flours and a multitude of unrecognized variables to name a few. What are the baseline and threshold values? The baseline and threshold values are used to exclude or filter out low intensity, poor-quality data before analysis. Baseline value is a number (such as 200, 500) that you set to filter out low intensity spots. For a spot with signal intensities in both channels below the user defined baseline value (before background subtraction), this spot will be be filtered out (filtering) leaving those features with signal greater than baseline value in at least one channel. A spot whose intensities in only one channel is below the user defined baseline value (before background subtraction) will be increased to this baseline value (scaling). The threshold value (T-value) is also a number (such as 1, 2, or 3) that you can set to filter out "noisy" spots. Based on the T-value, GPAP will calculate a dynamic value for each spot (Feature Background Intensity + [T-value * Background Standard Deviation]) and spots with intensities below the threshold in both channels (before background subtraction) will be filtered out. How do I determine an appropriate baseline and threshold value? You should set these two criteria to filter and scale the raw intensity data in GRP files appropriately. We recommend you begin with the default values (baseline value of 200 and not threshold value) along with an appropriate normalization method and view the resulting "before and after" diagnostic plots to evaluate the impact of these settings. Currently, the baseline cannot be set to a value below 200 and we suggest that you use at least the minimum baseline value. Hybridizations with extremely high background and noise will require more stringent filtering than the default. Which normalization method should I choose? If your data are not already normalized, we recommend a non-linear normalization method which is based on feature intensity (Loess - Global intensity dependent normalization) and/or spatial grouping (Loess Within-print-tip-group intensity dependent normalization). These non-linear normalization methods are widely accepted and applicable to most microarray data. Additional normalization methods (Linear Global Median, etc) are available if needed or warranted. Evaluation of the resulting "before and after" diagnostic plots will help determine if your data are better suited for one normalization method over another. For example, the distribution of log-ratios as seen in the Box Plots for individual arrays may reveal a spatial effect indicating "Loess - Within-print-tip-group intensity dependent normalization" is warranted. (NOTE: Remember, normalization is used to correct for systematic variation NOT biological variation. Sub-genomics arrays with few probes, probes selected with bias for participation in your treatment (subtracted cDNA libraries, SSH libraries, selected oligo subsets, etc) and/or treatments creating global changes (e.g., starvation, heatshock, etc) may alter the expression of so many features on your array that these normalization methods could obscure true changes in expression. Reliable normalization controls (such as heterologous genes and companion RNA spikes) are required in these cases and normalization to those controls should be done in GenePix Pro with no additional normalization applied in GPAP.) What is an outlier? An outlier is defined as a value far from most others in a set of data. An outlier that is many standard deviations from the mean will have dramatic impact on the average. Outliers should be identified and cast out of the data set to obtain a more meaningful average. However, identifying a large number of outliers for a given gene can indicate true variability of the mRNA abundance - an equally valuable measurement. The outlier definition is a number (such as 1, 2 or 3) used by GPAP to identify and remove outliers to calculate the final average Log2 ratios within and across array's replicates for each gene and is included in the Gene Summary Report . If the outlier definition is set at "2", any Log2 ratio that is outside of 2 standard deviations of the mean is considered as outlier, is removed and the average is re-calculated. If the outlier definition is set at "3", any Log2 ratio that is outside of 3 standard deviations of the mean is considered as outlier, is removed and the average is re-calculated. What are box plots? A box plot is a plot represents graphically several descriptive statistics of a given data set, which usually has a box including a central line and two tails. The the upper and lower boundary of the box show the location of the 75th percentile and 25th quartile respectively. The median, or central 50% of the data is drawn inside the box and the central line in the box shows the position of the median. The lines extending from the box disply the spread of the data. Replicate arrays must be similar in range or otherwise should be discarded. Box plots are provided for all of the data points in the GPRs and for each print-tip-group within individual GPRs and are drawn for the raw data and the processed (filtered and normalized) data. Box plots for individual arrays are helpful in diagnosing spatial effects and observing the impact of "Loess Within-print-tip-group intensity dependent normalization". Thus, box plots can be useful for visually comparing different normalization methods. Box plots are produced using the R statistical language with the Bioconductor package. What are scatter plots? The scatter plot is one of the simplest methods used to visualize overall mRNA expression levels within a single hybridization. The M-A scatter plot is a convenient to observe the distribution of intensity values and log ratios. M-A scatter plots are provided for each array and for separate blocks of each array. The colored lines appearing within the scatter plot represent the average for each print-tip-group and are drawn for the raw data and the processed (filtered and normalized) data. The M-A scatter plot is a Log2ratio (log2(cy5/cy3) ) vs. log2 intensity (1/2(log2(cy5*cy3))) plot. Therefore, if a gene has equal expression values in both the control and experiment, the expression ratio ( log2 ratio) will be zero. For a typical hybridization experiment, most genes will have equal expression values in both control and experiment and we expect the majority of points to be grouped around the horizontal line Y=0. Without normalization, the majority of points may be clustered around a horizontal line greater or less than Y=0 indicating the need for normalization. Notice that unrealistic and often dramatic scattering of data points is often observed prior to processing as "A" or log2 intensity (log2(cy5*cy3)) approaches zero, emphasizing the need to filter out low intensity, poor-quality data before analysis and interpretation. The baseline and/or threshold values should be stringent enough to remove the scattering near the lower intensities and increase the reliability of the reported ratios. Scatter plots are produced using the R statistical language with the Bioconductor package. What is a Q-Q plot? The Q-Q (Quantile-Quantile) plot provides a visual comparison of two populations and is a plot of the sampled t-statistic vs. a theoretical t-statistic. The Q-Q plot can indicate the degree a sample diverges from a normal distribution. Points which deviate markedly from a linear relationship to a theoretical t-statistic could be considered suspect genes exhibiting differentially expression. The Q-Q plots allow you to visualize the magnitude of differentially gene expression within the sample tested based on the Students ttest. However, the ordinary Student t-test is not ideally suited for microarray data because a large t-statistic can be driven by an unrealistically small standard deviation. The Q-Q plots are produced using the R statistical language with the Bioconductor package. What is a density plot? The density plots (density vs log2 intensity (1/2(log2(cy5*cy3)))) are of single-channel log-intensity densities and illustrate the distribution of single-channel intensities. Normally the distribution of intensities should appear roughly bell-shaped; however depending on the choice of genes and the experiment conducted, the distribution of intensities may appear double-peaked or skewed to one side. Density plots can be useful for visually comparing different normalization methods. Density plots are produced using the R statistical language with the LIMMA package. What is the B-statistic and how do I use it? The B-statistic is based on the Empirical Bayes approach to rank genes and determine if a gene is statistically significantly differential expressed or not. Classically, inference of significant changes in gene expression was based on a fixed value or absolute 2-fold or greater change (Log2 ratio >1 or <-1). However, this is an arbitrary threshold which can lead to false positive and false negative inferences and does not account for more subtle variations with biological significance. More recent approaches for determining significant changes in expression rely heavily on adequate biological replicate hybridizations and the calculation of a suitable statistic such as a moderated t-statistic or a b-statistic which ranks each gene to indicate whether a gene has significantly changed in expression or not. The ordinary t-statistic is not ideal because a large t-statistic can be driven by an unrealistically small standard deviation. An added advantage of the t-statistic is the introduction of standard deviation, number of replicates and sample variance to the averaged Log ratio. B-statistic is the log-odds that gene is differentially expressed. For example if B=1.5, the odds of differential expression is exp(1.5)=4.48, the probability that a gene is differentially expressed is 4.48/(4.48+1)=0.82, so there is 82% chance that gene is differentially expressed. The B-statistics is automatically adjusted for multiple testing by assuming that 1% genes are expected to be differentially expressed. In GPAP, the genes are ranked or scored according to the B-statistic and selection of a cut-off value is then determined by the user/investigator. If the B-statistic is 0 (zero), there is a 50% chance the measured Log2 ratio is random and not significant. The introduction of additional biological replicates (with high correlation coefficients) tends to produce higher B-statistic values. The higher the Bstatistic, the more significant the result. B-statistic, t-statistic and P-value (probability) are generated using the R statistical language with Bioconductor and the LIMMA package. What is M? M is the log-transformed ratio typically calculated as log2(cy5/cy3) or log2(treatment/control) and use used to instead of intensity ratios so the up-regulated and down-regulated values are of the same scale and comparable. A two-fold change is represented by a log2 ratio of 1.0 (up-regulation) or -1.0 (downregulation). A three-fold change in gene expression is represented by a Log2 Ratio of 1.58 or -1.58. This value is not averaged in the "M and A value for individual spots" report nor in the diagnostic plots. However, the M value is averaged in the "Gene Summary (averaged)" and "B-Statistics Ranking" reports for the same spot between slides and within slides if replicates are evenly printed on the array. What is A? A is typically referred to as the intensity or log2intensity and is calculated as 1/2(log2(cy5*cy3)). This value is useful to observe the distribution of signal intensities and to recognize if a spot produced brilliant or weak signal. Note that the signal intensities are the result of transcript abundance AND probe abundance, spot quality, hybridization kinetics, cross-hybridization events, etc thus a weak signal cannot be interpreted as low mRNA levels because the spot may simply be a bad probe!!!. This value is not averaged in the "M and A value for individual spots" nor in the diagnostic plots. However, the M value is averaged in the "Gene Summary (averaged)" and "B-Statistics Ranking" reports between slides and within slides if replicates are evenly printed on the array. What is t? "t" is a moderated t-statistic and is a ratio of Log2-expression level to its standard error. Moderated tstatistic has the same interpretation as an student t-statistic except that standard error have been moderated across genes, effectively borrowing information from the ensemble of genes to aid with inference about each individual gene. Moderated t-statistic and associated p-value do not require prior guess for the number of differntially expressed genes. What is P-value? The P-value is obtained from moderated t-statistic and after FDR adjustment which is Benjamini and Hochberg's method to control the false discovery rate. If you select all the genes with p-value less than a given value, say 0.05, as differentially expressed, then the expected proportion of false discovery in the selected group should be less than that value, in this case less than 5%. Among the three statistics, moderated-t, associated p-value and B-statistic, we usually base our gene select on p-value. The p-value will represent an area under a probability curve which is less than or greater than a significance level. The significance level is defined by the user and is normally 0.05 or less and and any P-values below that mark are considered "significant". P-values do not simple provide you with a "Yes" or "No" answer, they provide a sense of the strength based on the evidence. The lower the p-value, the stronger the evidence. What is SD? SD is the standard deviation of the log2 ratios for each gene. SD is one of several indices of variability used to characterize the dispersion among the measures in a given population. The standard deviation is the square root of the variance and is calculated as "sqroot [sum( x - u)^2 / (N-1)] where x is the log2 ratio of individual spots, u is the mean log2 ratio and N is the total number of spots. Variance is a measure of how spread out a distribution is. The standard deviation is a measure of how dispersed the measured values are from the mean. What is CV? CV or coefficient of variation is a statistic used to describe the amount of variation within a set of measurements and is calculated as the (SD of Log2 Ratios)/(Mean of Log2 Ratios). What is Weight? Weight is used in normalization. In normalization, good spots with high intensity in both channel are given full weight (weight equal 1) and bad spots which are flagged in GenPix or have low intensity in both channels are given low weight (weight equal 0.1). What is Fold Change? The Fold Change is calculated by the following formula: Fold change=2^(signal Log2 ratio) (signal log2 ratio>=0) Fold change=(-1)*2^(-1*signal Log2 ratio) (signal log2 ratio<0)