
25_DetailLectOutjk_AR
... The principle of maximum likelihood states that, given certain rules about how DNA changes over time, a tree should reflect the most likely sequence of evolutionary events. Maximum likelihood methods are designed to use as much information as possible. ...
... The principle of maximum likelihood states that, given certain rules about how DNA changes over time, a tree should reflect the most likely sequence of evolutionary events. Maximum likelihood methods are designed to use as much information as possible. ...

PPTX - Tandy Warnow
... How well do POY and BeeTLe do, compared to other MSA methods? • We simulated sequences down evolutionary trees with substitutions, insertions, and indels. • We computed alignments on each dataset using multiple techniques (e.g., POY, BeeTLe, Muscle, ...
... How well do POY and BeeTLe do, compared to other MSA methods? • We simulated sequences down evolutionary trees with substitutions, insertions, and indels. • We computed alignments on each dataset using multiple techniques (e.g., POY, BeeTLe, Muscle, ...



HW 10 key
... have the insertion. In this case, the gene tree will not match the species tree. The second method is lateral gene transfer. Any gene flow between closely related species after divergence can obscure the true species tree. For example, hybridization between two closely related daughter species can t ...
... have the insertion. In this case, the gene tree will not match the species tree. The second method is lateral gene transfer. Any gene flow between closely related species after divergence can obscure the true species tree. For example, hybridization between two closely related daughter species can t ...


Species Trees
... • Would need priors on hybridization and related phenomena • Computationally challenging (but progress being made) • Assumes that individual gene tree estimates are valid • A reticulogram is not very useful for classification ...
... • Would need priors on hybridization and related phenomena • Computationally challenging (but progress being made) • Assumes that individual gene tree estimates are valid • A reticulogram is not very useful for classification ...

Many of the slides that I`ll use have been borrowed from Dr. Paul
... When we create a character matrix for Hennig’s system, it is crucial that: • traits assigned the same state represent homologous states (trace back to the MRCA) • we correctly identify the directionality of the transformations (which state is plesiomorphic and which is apomorphic). The process of id ...
... When we create a character matrix for Hennig’s system, it is crucial that: • traits assigned the same state represent homologous states (trace back to the MRCA) • we correctly identify the directionality of the transformations (which state is plesiomorphic and which is apomorphic). The process of id ...

Is the Tiger a Copycat? A Phylogenetic Analysis Laboratory
... only partial segments of the gene being analyzed. In order to maximize the likelihood of a successful alignment, excess sequence data was trimmed from the beginning and end of some of the FASTA sequences. This ensured that the alignment software was only matching up homologous segments of the genes ...
... only partial segments of the gene being analyzed. In order to maximize the likelihood of a successful alignment, excess sequence data was trimmed from the beginning and end of some of the FASTA sequences. This ensured that the alignment software was only matching up homologous segments of the genes ...

A Genetic Algorithm for Maximum-Likelihood Phylogeny Inference
... Phylogeny reconstruction is a difficult computational problem, because the number of possible solutions increases with the number of included taxa. For example, for only 14 taxa, there are more than seven trillion possible unrooted phylogenetic trees. For this reason, phylogenetic inference methods ...
... Phylogeny reconstruction is a difficult computational problem, because the number of possible solutions increases with the number of included taxa. For example, for only 14 taxa, there are more than seven trillion possible unrooted phylogenetic trees. For this reason, phylogenetic inference methods ...

Align the DNA sequences
... With a gap correctly inserted, it is now apparent that the two organisms share 11 of the 12 bases being examined (92% sequence homology). Correct alignment is difficult and usually done through the use of software such as CLUSTAL. Step 3: Construct a Phylogenetic Tree- With the sequences correctly a ...
... With a gap correctly inserted, it is now apparent that the two organisms share 11 of the 12 bases being examined (92% sequence homology). Correct alignment is difficult and usually done through the use of software such as CLUSTAL. Step 3: Construct a Phylogenetic Tree- With the sequences correctly a ...

Protein Evolution and Sequence Analysis
... frequently inserting into new locations with different expression patterns. The mechanism by which new genes/proteins arise allow for the possibility of sequence analysis to infer functional and structural relationships among different sequences. ...
... frequently inserting into new locations with different expression patterns. The mechanism by which new genes/proteins arise allow for the possibility of sequence analysis to infer functional and structural relationships among different sequences. ...

Evolutionary dynamics and emergence of panzootic H5N1 Influenza
... Models of nucleotide evolution Several probabilistic models of evolution have been developed to convert observed nucleotide distances into measures of actual evolutionary distances The relative complexity of these models is a function of the extent of the biological, biochemical ad evolutionary ass ...
... Models of nucleotide evolution Several probabilistic models of evolution have been developed to convert observed nucleotide distances into measures of actual evolutionary distances The relative complexity of these models is a function of the extent of the biological, biochemical ad evolutionary ass ...


phylogeny and evolution
... the species. By comparing the similarity of DNA between two species, scientists can determine how closely they are related. These molecular similarities reveal the relationships among organisms. The study of ancestral relations among species, often illustrated with a "tree of life" branching diagram ...
... the species. By comparing the similarity of DNA between two species, scientists can determine how closely they are related. These molecular similarities reveal the relationships among organisms. The study of ancestral relations among species, often illustrated with a "tree of life" branching diagram ...



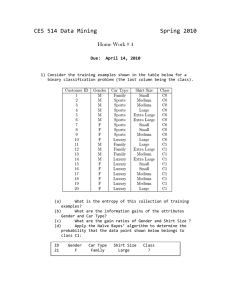
CES 514 Data Mining Fall 2003
... presented in class to determine the class to which the above data point belongs. 2) You are given a data set that contains various attributes. The last column contains the classification (0 or 1). (a) Using Weka, create a decision tree based on 80% of the data points and apply it to the remaining 20 ...
... presented in class to determine the class to which the above data point belongs. 2) You are given a data set that contains various attributes. The last column contains the classification (0 or 1). (a) Using Weka, create a decision tree based on 80% of the data points and apply it to the remaining 20 ...

Lecture-TreeOfLife
... uncertainty about the actual tree topology (nd, not determined). (B. aphidicola strains are entirely isolated in different hosts and were thus considered as different species despite having a single name. In B. aphidicola, amounts of gene loss and gene gain are similar, suggesting that LGT is overes ...
... uncertainty about the actual tree topology (nd, not determined). (B. aphidicola strains are entirely isolated in different hosts and were thus considered as different species despite having a single name. In B. aphidicola, amounts of gene loss and gene gain are similar, suggesting that LGT is overes ...

Title Pruning Decision Trees Using Rules3 Inductive Learning
... Induction, Inductive Learning, Decision Tress, Pruning. One important disadvantage of decision tree based inductive learning algorithms is that they use some irrelevant values to establish the decision tree. This causes the final rule set to be less general. To overcome with this problem the tree ha ...
... Induction, Inductive Learning, Decision Tress, Pruning. One important disadvantage of decision tree based inductive learning algorithms is that they use some irrelevant values to establish the decision tree. This causes the final rule set to be less general. To overcome with this problem the tree ha ...

