
Maximum pseudo-likelihood estimation of species trees (MP
... The program is written in C. To compile the source code, type “make” (without quote) at the terminal window on Mac (or the Dos command window on PC). You need to set “architecture = ?” to the correct platform (mac, unix, or windows) in makefile. Currently, the parallel version is not available. Thus ...
... The program is written in C. To compile the source code, type “make” (without quote) at the terminal window on Mac (or the Dos command window on PC). You need to set “architecture = ?” to the correct platform (mac, unix, or windows) in makefile. Currently, the parallel version is not available. Thus ...

Trees
... Amino-acid sites are partially ordered characters. An amino acid cannot change into all other amino acids in a singe step, as sometimes 2 or 3 steps are required. For example, a tyrosine may only change into a leucine through an intermediate state, i.e., phenylalanine or histidine. ...
... Amino-acid sites are partially ordered characters. An amino acid cannot change into all other amino acids in a singe step, as sometimes 2 or 3 steps are required. For example, a tyrosine may only change into a leucine through an intermediate state, i.e., phenylalanine or histidine. ...





2_Outline_BIO119_div..
... B. Example: Genus, Species: Escherichia coli must be Latin endings. 1. Genus is always capitalized and the species is lower case 2. Always italicize or underline. 3. Name usually has some significance. C. How do identify a new isolate and classify it to the species level? 1. There are international ...
... B. Example: Genus, Species: Escherichia coli must be Latin endings. 1. Genus is always capitalized and the species is lower case 2. Always italicize or underline. 3. Name usually has some significance. C. How do identify a new isolate and classify it to the species level? 1. There are international ...




What is Bioinformatics I?
... Phylogenetic analysis of molecular sequences with an emphasis on methods of phylogenetic inference and hypothesis testing. Gene and genome history, gene family evolution, inference of ancestral proteins, and phylogenetic analysis as a predictive tool. (3 weeks) ...
... Phylogenetic analysis of molecular sequences with an emphasis on methods of phylogenetic inference and hypothesis testing. Gene and genome history, gene family evolution, inference of ancestral proteins, and phylogenetic analysis as a predictive tool. (3 weeks) ...


Slajd 1
... The construction of phylogenetic trees from numerical methods The principle of maximum parsimony (Occam’s razor) holds that we should accept that phylogenetic tree that can be constructed with the least number of morphological changes. The raw data ...
... The construction of phylogenetic trees from numerical methods The principle of maximum parsimony (Occam’s razor) holds that we should accept that phylogenetic tree that can be constructed with the least number of morphological changes. The raw data ...

here - carrot!!!
... Exploration of the tree space by sampling trees using a biased random walk (Implemented in MrBayes program) Trees with higher likelihoods will be sampled more often ...
... Exploration of the tree space by sampling trees using a biased random walk (Implemented in MrBayes program) Trees with higher likelihoods will be sampled more often ...

Document
... • An assumption of the algorithm is that the molecular clock is constant for sequences in the tree. If there are unequal substitution rates, the tree may be wrong. • While UPGMA is simple, it is less accurate than the neighbor-joining approach (described next). ...
... • An assumption of the algorithm is that the molecular clock is constant for sequences in the tree. If there are unequal substitution rates, the tree may be wrong. • While UPGMA is simple, it is less accurate than the neighbor-joining approach (described next). ...



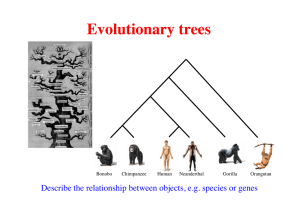
phylogenetic tree.
... Its tools include fossils, morphology, genes, and molecular evidence. Taxonomy is an ordered division of organisms into categories based on a set of characteristics used to assess similarities and differences. Binomial nomenclature uses a two-part naming system that consists of the genus to which th ...
... Its tools include fossils, morphology, genes, and molecular evidence. Taxonomy is an ordered division of organisms into categories based on a set of characteristics used to assess similarities and differences. Binomial nomenclature uses a two-part naming system that consists of the genus to which th ...
File - Down the Rabbit Hole
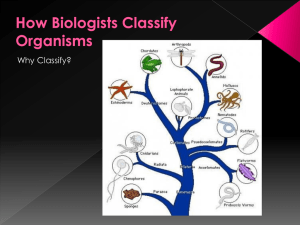
... nested groups, in which similar or related groups at one level are combined into larger and more general groups at the next higher level. Biological classification is based on shared descent from the nearest common ancestor ...
... nested groups, in which similar or related groups at one level are combined into larger and more general groups at the next higher level. Biological classification is based on shared descent from the nearest common ancestor ...

Shared character
... into thea diff charactersitsica – derived.. between species by making a tree Just by looking at the amino acid sequences, biologist can find relationships characteristics that makes it differ Molecular cladogram- branch lengths proportional to #of amino acid changes Shared characterists – the ones t ...
... into thea diff charactersitsica – derived.. between species by making a tree Just by looking at the amino acid sequences, biologist can find relationships characteristics that makes it differ Molecular cladogram- branch lengths proportional to #of amino acid changes Shared characterists – the ones t ...

Introduction into Phylogenetics I Introduction: A. Phylogenies depict
... we place it next to the gorilla, who also does not have a tail. But if we do this, we are making 5 assumptions of gained or lost characteristics. G. If we put the frog next to the salamander and lizard, we only have to make 4 assumptions, which is more simple, so we go with the left Phylogenetic tre ...
... we place it next to the gorilla, who also does not have a tail. But if we do this, we are making 5 assumptions of gained or lost characteristics. G. If we put the frog next to the salamander and lizard, we only have to make 4 assumptions, which is more simple, so we go with the left Phylogenetic tre ...

