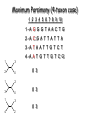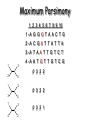Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Point mutation wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Koinophilia wikipedia , lookup
Metagenomics wikipedia , lookup
Gene expression programming wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Microevolution wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
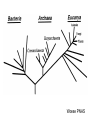
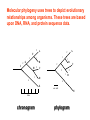
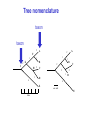
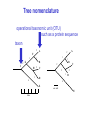
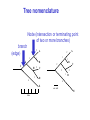
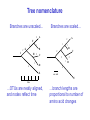
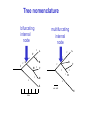
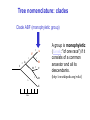
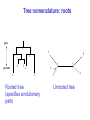
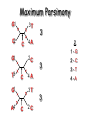
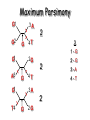
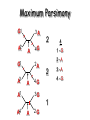
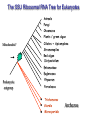
Molecular Phylogenetics Introduction to evolution and phylogeny Nomenclature of trees Four stages of molecular phylogeny: [1] selecting sequences [2] multiple sequence alignment [3] tree-building [4] tree evaluation Practical approaches to making trees Introduction At the molecular level, evolution is a process of mutation with selection. Molecular evolution is the study of changes in genes and proteins throughout different branches of the tree of life. Phylogeny is the inference of evolutionary relationships. Traditionally, phylogeny relied on the comparison of morphological features between organisms. Today, molecular sequence data are also used for phylogenetic analyses. corrected amino acid changes per 100 residues (m) Dickerson (1971) Millions of years since divergence Molecular clock for proteins: rate of substitutions per aa site per 109 years Fibrinopeptides Kappa casein Lactalbumin Serum albumin Lysozyme Trypsin Insulin Cytochrome c Histone H2B Ubiquitin Histone H4 9.0 3.3 2.7 1.9 0.98 0.59 0.44 0.22 0.09 0.010 0.010 Molecular clock hypothesis: implications If protein sequences evolve at constant rates, they can be used to estimate the times that sequences diverged. This is analogous to dating geological specimens by radioactive decay. Molecular clock hypothesis: implications If protein sequences evolve at constant rates, they can be used to estimate the times that sequences diverged. This is analogous to dating geological specimens by radioactive decay. N = total number of substitutions L = number of nucleotide sites compared between two sequences K= N L = number of substitutions per nucleotide site Rate of nucleotide substitution r and time of divergence T r = rate of substitution = 0.56 x 10-9 per site per year for hemoglobin alpha K = 0.093 = number of substitutions per nucleotide site (rat versus human) r = K / 2T T = .093 / (2)(0.56 x 10-9) = 80 million years Neutral theory of evolution An often-held view of evolution is that just as organisms propagate through natural selection, so also DNA and protein molecules are selected for. According to Motoo Kimura’s 1968 neutral theory of molecular evolution, the vast majority of DNA changes are not selected for in a Darwinian sense. The main cause of evolutionary change is random drift of mutant alleles that are selectively neutral (or nearly neutral). Positive Darwinian selection does occur, but it has a limited role. Goals of molecular phylogeny Phylogeny can answer questions such as: • How many genes are related to my favorite gene? • Was the extinct quagga more like a zebra or a horse? • Was Darwin correct that humans are closest to chimps and gorillas? • How related are whales, dolphins & porpoises to cows? • Where and when did HIV originate? • What is the history of life on earth? Woese PNAS Molecular phylogeny: nomenclature of trees There are two main kinds of information inherent to any tree: topology and branch lengths. We will now describe the parts of a tree. Molecular phylogeny uses trees to depict evolutionary relationships among organisms. These trees are based upon DNA, RNA, and protein sequence data. 2 A 1 I 2 1 1 G B H 2 1 6 1 2 C 2 D B C 2 1 E A 2 F D 6 one unit E time chronogram phylogram Tree nomenclature taxon taxon 2 A 1 I 2 1 1 G B H 2 1 6 1 2 C 2 D B C 2 1 E A 2 F D 6 one unit E time Tree nomenclature operational taxonomic unit (OTU) such as a protein sequence taxon 2 A 1 I 2 1 1 G B H 2 1 6 1 2 C 2 D B C 2 1 E A 2 F D 6 one unit E time Tree nomenclature Node (intersection or terminating point of two or more branches) branch (edge) 2 I 1 1 G B H 2 1 6 1 2 C 2 D B C 2 1 E A 2 F 1 2 A D 6 one unit E time Tree nomenclature Branches are unscaled... 2 Branches are scaled... A 1 I 2 1 1 G B H 2 1 6 1 2 C 2 D B C 2 1 E A 2 F D 6 one unit E time …OTUs are neatly aligned, and nodes reflect time …branch lengths are proportional to number of amino acid changes Tree nomenclature bifurcating internal node multifurcating internal node 2 A 1 I 2 1 1 G B H 2 1 6 A 2 F B 2 C 2 2 1 D E C D 6 one unit E time Examples of multifurcation: failure to resolve the branching order of some metazoans and protostomes Rokas A. et al., Animal Evolution and the Molecular Signature of Radiations Compressed in Time, Science 310:1933, 23 December 2005, Fig. 1. Tree nomenclature: clades Clade ABF (monophyletic group) 2 F 1 I 2 A 1 B G H 2 1 6 C D E time A group is monophyletic (Greek: "of one race") if it consists of a common ancestor and all its descendants. (http://en.wikipedia.org/wiki/) Tree roots The root of a phylogenetic tree represents the common ancestor of the sequences. Some trees are unrooted, and thus do not specify the common ancestor. A tree can be rooted using an outgroup (that is, a taxon known to be distantly related from all other OTUs). Tree nomenclature: roots past 9 1 7 5 8 6 2 present 1 7 3 4 2 5 Rooted tree (specifies evolutionary path) 8 6 3 Unrooted tree 4 Tree nomenclature: outgroup rooting past root 9 10 7 8 7 6 2 present 9 8 3 4 1 Rooted tree 2 5 1 3 4 5 6 Outgroup (used to place the root) Enumerating trees Cavalii-Sforza and Edwards (1967) derived the number of possible unrooted trees (NU) for n OTUs (n > 3): NU = (2n-5)! 2n-3(n-3)! The number of bifurcating rooted trees (NR) (2n-3)! NR = n-2 2 (n-2)! For 10 OTUs (e.g. 10 DNA or protein sequences), the number of possible rooted trees is 34 million, and the number of unrooted trees is 2 million. Many tree-making algorithms can exhaustively examine every possible tree for up to ten to twelve sequences. Species trees versus gene/protein trees Molecular evolutionary studies can be complicated by the fact that both species and genes evolve. speciation usually occurs when a species becomes reproductively isolated. In a species tree, each internal node represents a speciation event. Genes (and proteins) may duplicate or otherwise evolve before or after any given speciation event. The topology of a gene (or protein) based tree may differ from the topology of a species tree. Species trees versus gene/protein trees past speciation event present species 1 species 2 Species trees versus gene/protein trees Gene duplication events species 1 speciation event species 2 Species trees versus gene/protein trees Gene duplication events speciation event OTUs species 1 species 2 Orthology/paralogy Orthologous genes are homologous (corresponding) genes in different species (genomes) Paralogous genes are homologous genes within the same species (genome) Four stages of phylogenetic analysis Molecular phylogenetic analysis may be described in four stages: [1] Selection of sequences for analysis [2] Multiple sequence alignment [3] Tree building [4] Tree evaluation Stage 2: Multiple sequence alignment The fundamental basis of a phylogenetic tree is a multiple sequence alignment. (If there is a misalignment, or if a nonhomologous sequence is included in the alignment, it will still be possible to generate a tree.) Two Major Approaches to Phylogeny Inference 1) Distance Matrix Methods Calculate matrix of pairwise distances from all data, then infer tree using a clustering algorithm. 2) Character Based Methods (maximum parsimony) Inspect columns of characters, infer trees from columns that contain “informative” characters, and use these to infer most likely tree given the data. Distance Matrix Methods (matrix calculation) Reality: Not all sites are free to change, the same sites change multiple times The simplest model is that of Jukes & Cantor Jukes & Cantor: dxy = -(3/4) ln (1-4/3 D) • dxy = distance between sequence x and sequence y expressed as the number of changes per site • (note dxy = r/n where r is number of replacements and n is the total number of sites. This assumes all sites can vary and when unvaried sites are present in two sequences it will underestimate the amount of change which has occurred at variable sites) (i.e., previous reality check) • D = is the observed proportion of nucleotides which differ between two sequences (fractional dissimilarity) • ln = natural log function to correct for superimposed substitutions (in general logging tends to convert exponential trends to linear trends) • The 3/4 and 4/3 terms reflect that there are four types of nucleotides and three ways in which a second nucleotide may not match a first - with all types of change being equally likely (i.e. unrelated sequences should be 25% identical by chance alone) The natural logarithm ln is used to correct for superimposed changes at the same site • • • • If two sequences are 95% identical they are different at 5% or 0.05 (D) of sites thus: – dxy = -3/4 ln (1-4/3 0.05) = 0.0517 Note that the observed dissimilarity 0.05 increases only slightly to an estimated 0.0517 - this makes sense because in two very similar sequences one would expect very few changes to have been superimposed at the same site in the short time since the sequences diverged apart However, if two sequences are only 50% identical they are different at 50% or 0.50 (D) of sites thus: – dxy = -3/4 ln (1-4/3 0.5) = 0.824 For dissimilar sequences, which may diverged apart a long time ago, the use of ln infers that a much larger number of superimposed changes have occurred at the same site Distance Matrix Methods (tree construction) UPGMA is unweighted pair group method using arithmetic mean 1 2 3 4 5 Tree-building methods: UPGMA Step 1: compute the pairwise distances of all the proteins. Get ready to put the numbers 1-5 at the bottom of your new tree. 1 2 3 4 5 Tree-building methods: UPGMA Step 2: Find the two proteins with the smallest pairwise distance. Cluster them. 1 2 6 3 4 5 1 2 Tree-building methods: UPGMA Step 3: Do it again. Find the next two proteins with the smallest pairwise distance. Cluster them. 1 2 6 1 3 4 5 7 2 4 5 Tree-building methods: UPGMA Step 4: Keep going. Cluster. 1 8 2 7 6 3 4 5 1 2 4 5 3 Tree-building methods: UPGMA Step 4: Last cluster! This is your tree. 9 1 2 8 7 3 6 4 5 1 2 4 5 3 Distance-based methods: UPGMA trees UPGMA is a simple approach for making trees. • An UPGMA tree is always rooted. • An assumption of the algorithm is that the molecular clock is constant for sequences in the tree. If there are unequal substitution rates, the tree may be wrong. • While UPGMA is simple, it is less accurate than the neighbor-joining approach (described next). Distance method: Advantages • Fast - suitable for analysing data sets which are too large for other more computationally intensive methods such as maximum likelihood • A large number of models are available with many parameters improves estimation of distances Distance method: Disadvantages • Information is lost - given only the distances, it is impossible to derive the original sequences • Only through character based analyses can the history of sites be investigated; e.g., most informative positions be inferred Character Based Methods: Maximum Parsimony The best tree: should be the one that requires the smallest number of substitutions to explain the differences among the sequences being studied. Occam's razor: Among his statements (translated from his Latin) are: "Plurality is not to be assumed without necessity" and "What can be done with fewer [assumptions] is done in vain with more." One consequence of this methodology is the idea that the simplest or most obvious explanation of several competing ones is the one that should be preferred until it is proven wrong. Not all Characters are Used in Parsimony Analysis • informative sites - nucleotide (or amino acid) columns that are represented by at least two different character states found in at least two different sequences, these sites allow the distinction between alternative trees. • uninformative sites - nucleotide (or amino acid) columns that do not allow the distinction between two trees (e.g., constant) Maximum Parsimony (4-taxon case) 1 2 3 4 5 6 7 8 9 10 1-A G G G T A A C T G 2-A C G A T T A T T A 3-A T A A T T G T C T 4-A A T G T T G T C G 1 3 2 4 1 2 3 4 1 3 4 2 How may informative sites are there in this data set? Maximum Parsimony (4-taxon case) 1 2 3 4 5 6 7 8 9 10 1-A G G G T A A C T G 2-A C G A T T A T T A 3-A T A A T T G T C T 4-A A T G T T G T C G 1 3 2 4 1 2 3 4 1 3 0 3 0 3 0 3 4 2 Maximum Parsimony G1 C2 3T C 3 4A 2 1-G G1 T3 2C C G1 3 4A 3T C 2C 3-T 4-A 3 A4 2-C Maximum Parsimony 1 2 3 4 5 6 7 8 9 10 1-A G G G T A A C T G 2-A C G A T T A T T A 3-A T A A T T G T C T 4-A A T G T T G T C G 1 3 2 4 1 2 3 4 1 3 0 3 2 0 3 2 0 3 2 4 2 Maximum Parsimony G1 G2 3A G 2 4T 3 1-G G1 A3 2G G G1 2-G 2 4T 4-T 3A 2 T4 G 2G 3-A Maximum Parsimony 1 2 3 4 5 6 7 8 9 10 1-A G G G T A A C T G 2-A C G A T T A T T A 3-A T A A T T G T C T 1 4-A A T G T T G T C G 3 0 3 2 2 2 4 1 2 0 3 2 2 3 4 1 3 4 2 0 3 2 1 Maximum Parsimony G1 A2 3A A G1 A3 4G 2A A A1 2 1-G 2-A 2 4G 3G 1 A4 A 2G 4 3-A 4-G Maximum Parsimony 1 3 2 4 1 2 3 4 1 3 0 3 2 2 0 1 1 1 1 3 14 0 3 2 2 0 1 2 1 2 3 16 0 3 2 1 0 1 2 1 2 3 15 4 2 Maximum Parsimony 1 3 2 4 1 2 3 4 5 6 7 8 9 10 1-A G G G T A A C T G 2-A C G A T T A T T A 3-A T A A T T G T C T 4-A A T G T T G T C G 0 3 2 2 0 1 1 1 1 3 14 Parsimony - advantages • is a simple method - easily understood operation • does not seem to depend on an explicit model of evolution • gives both trees and associated hypotheses of character evolution • should give reliable results if the data is well structured and homoplasy is either rare or widely (randomly) distributed on the tree Parsimony - disadvantages • May give misleading results if homoplasy is common or concentrated in particular parts of the tree, e.g: - thermophilic convergence - base composition biases - long branch attraction • Underestimates branch lengths (Why?) • Model of evolution is implicit - behaviour of method not well understood • Parsimony often justified on purely philosophical grounds - we must prefer simplest hypotheses - particularly by morphologists • For most molecular systematists, this is uncompelling Parsimony can be inconsistent • • Felsenstein (1978) developed a simple model phylogeny including four taxa and a mixture of short and long branches Under this model parsimony will give the wrong tree A B Model tree p p q C q q D Parsimony tree Rates or Branch lengths p >> q C A Wrong B D Long branches are attracted but the similarity is homoplastic • With more data the certainty that parsimony will give the wrong tree increases - so that parsimony is statistically inconsistent • Advocates of parsimony initially responded by claiming that Felsenstein’s result showed only that his model was unrealistic • It is now recognised that the long-branch attraction (in the “Felsenstein Zone”) is one of the most serious problems in phylogenetic inference Summary and recommendations • Remember that molecular phylogenetics yields gene trees • Accurate gene trees may not be accurate organismal trees • Gene duplications and paralogy, and lateral transfer can produce mismatches between gene and organismal phylogenies • Use congruence between separate gene trees to identify robust organismal phylogenies or mismatches that require further information The most famous case of LBA misleading biologists The Universal SSU rRNA Tree Wheelis et al. 1992 PNAS 89: 2930 The SSU Ribosomal RNA Tree for Eukaryotes Animals Fungi Choanozoa Plants / green algae Mitochondria? Ciliates + Apicomplexa Stramenopiles Red algae Dictyostelium Entamoebae Prokaryotic outgroup Euglenozoa Physarum Percolozoa Trichomonas Giardia Microsporidia Archezoa