
A Bayesian Method for Rank Agreggation
... ◦ j , the relative ranks within “targets”, i.e., with Di 1 ◦ R1|0, the relative ranks of each target among j the null genes. ...
... ◦ j , the relative ranks within “targets”, i.e., with Di 1 ◦ R1|0, the relative ranks of each target among j the null genes. ...


A Novel Method to Detect Identities in tRNA Genes Using Sequence
... We applied the method to Class I tRNAs to detect characteristic sites. We found that about 40% of characteristic sites that we detected are identities that have been detected experimentally, and that the remaining characteristic sites are in T and D domains which are the elbow regions of tRNAs. This ...
... We applied the method to Class I tRNAs to detect characteristic sites. We found that about 40% of characteristic sites that we detected are identities that have been detected experimentally, and that the remaining characteristic sites are in T and D domains which are the elbow regions of tRNAs. This ...

Text S1.
... Most of the analyses were carried out manually with the sequence editor ED [13]. Briefly, each sequence from [1,2] was visually compared with its most similar counterparts in order to detect frameshifts (leading to stretches of amino acids highly different from the consensus) and with all its homolo ...
... Most of the analyses were carried out manually with the sequence editor ED [13]. Briefly, each sequence from [1,2] was visually compared with its most similar counterparts in order to detect frameshifts (leading to stretches of amino acids highly different from the consensus) and with all its homolo ...

Notes 3
... Different genes evolve at different rates, which makes them useful for analyzing species that diverged at different times in the past. Ribosomal RNA evolves very slowly. The recognition that Archaea and Bacteria were quite different first came from the analysis of ribosomal RNA sequences. Once the g ...
... Different genes evolve at different rates, which makes them useful for analyzing species that diverged at different times in the past. Ribosomal RNA evolves very slowly. The recognition that Archaea and Bacteria were quite different first came from the analysis of ribosomal RNA sequences. Once the g ...


Math 7 Standards
... 7.SP.1-Understand that statistics can be used to gain information about a population by examining a sample. 7.SP.2-Use data from a random sample to draw inferences about a population with an unknown characteristic. 7.SP.3-Informally assess the degree of visual overlap of two numerical data distribut ...
... 7.SP.1-Understand that statistics can be used to gain information about a population by examining a sample. 7.SP.2-Use data from a random sample to draw inferences about a population with an unknown characteristic. 7.SP.3-Informally assess the degree of visual overlap of two numerical data distribut ...



presentation source
... a reward for character matches, a small penalty for character mismatches, and a large penalty for gaps. These penalties should be such that the highest scoring alignment is the most likely one to reflect the true evolutionary relationship of the loci. Two sequences can be considered “similar” if the ...
... a reward for character matches, a small penalty for character mismatches, and a large penalty for gaps. These penalties should be such that the highest scoring alignment is the most likely one to reflect the true evolutionary relationship of the loci. Two sequences can be considered “similar” if the ...

Document
... The bottom line of states are the main states (M) •These model the columns of the alignment The second row of diamond shaped states are called the insert states (I) •These are used to model the highly variable regions in the alignment. The top row or circles are delete states (D) •These are silent ...
... The bottom line of states are the main states (M) •These model the columns of the alignment The second row of diamond shaped states are called the insert states (I) •These are used to model the highly variable regions in the alignment. The top row or circles are delete states (D) •These are silent ...


attachment=1477
... CS2032 DATA WAREHOUSING AND DATA MINING Note: 1.When u study the dwdm..study these topics and then move to some other topics wat u feel as important 2.most of the theory questions during the valuation they wil see correct definitions,key points,sub headings,presentation.... 3.Dont mugup all the poin ...
... CS2032 DATA WAREHOUSING AND DATA MINING Note: 1.When u study the dwdm..study these topics and then move to some other topics wat u feel as important 2.most of the theory questions during the valuation they wil see correct definitions,key points,sub headings,presentation.... 3.Dont mugup all the poin ...

AP Biology Diversity Standards 1.A.1: Natural selection is a major
... biological processes and features shared by all domains or within one domain of life, and how these shared, conserved core processes and features support the concept of common ancestry for all organisms. ...
... biological processes and features shared by all domains or within one domain of life, and how these shared, conserved core processes and features support the concept of common ancestry for all organisms. ...
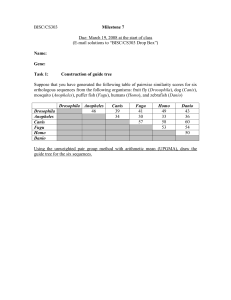
Milestone7
... phylogenies, and has greatly improved our phylogenetic knowledge. However care must be taken when constructing phylogenetic trees based on molecular data. Phylogenetic trees based on gene sequences do not always correspond exactly to the phylogenetic trees showing the evolutionary relationships betw ...
... phylogenies, and has greatly improved our phylogenetic knowledge. However care must be taken when constructing phylogenetic trees based on molecular data. Phylogenetic trees based on gene sequences do not always correspond exactly to the phylogenetic trees showing the evolutionary relationships betw ...

Understanding selectivity in the CRISPR CAS9 system
... be reduced to a minimum because its occurrence can lead to modifications of genes rather than the one effectively targeted, with unpredictable consequences. Hence, an important question is to understand what are the intrinsic limits in terms of targeting selectivity that such system must have. For e ...
... be reduced to a minimum because its occurrence can lead to modifications of genes rather than the one effectively targeted, with unpredictable consequences. Hence, an important question is to understand what are the intrinsic limits in terms of targeting selectivity that such system must have. For e ...

Lecture 6
... • Example: MSA • Topology—which sequences should be aligned first • Distance—how to weight the sequences when computing alignment score ...
... • Example: MSA • Topology—which sequences should be aligned first • Distance—how to weight the sequences when computing alignment score ...

Tuesday, March 24 - Perry Local Schools
... 10) A node is a place where a branch splits. It represents the most common ancestor by a clade. ...
... 10) A node is a place where a branch splits. It represents the most common ancestor by a clade. ...

COT6930 Course Project
... • Genes are considered independently. – Redundant genes may be included. – Some genes jointly with strong discriminant power but individually are weak will be ignored. • Good single features do not necessarily form a good feature set ...
... • Genes are considered independently. – Redundant genes may be included. – Some genes jointly with strong discriminant power but individually are weak will be ignored. • Good single features do not necessarily form a good feature set ...

PhyloPat2 - Department of Computing Science
... in a set of whole genome sequences Can be used to determine sets of genes that occur only in certain evolutionary branches More Common as increasing amounts of orthology data have become available Phylogenetic Patterns Search tools are available for querying proteins, but not for querying gene ...
... in a set of whole genome sequences Can be used to determine sets of genes that occur only in certain evolutionary branches More Common as increasing amounts of orthology data have become available Phylogenetic Patterns Search tools are available for querying proteins, but not for querying gene ...


Orthology, paralogy and GO annotation
... An “ortholog cluster” is made by one or more “slices” through the protein family tree Some combination of evolutionary rates and history of duplications Might miss genes that could be efficiently annotated at the same time From a strict evolutionary standpoint, orthologs are separated ONLY by specia ...
... An “ortholog cluster” is made by one or more “slices” through the protein family tree Some combination of evolutionary rates and history of duplications Might miss genes that could be efficiently annotated at the same time From a strict evolutionary standpoint, orthologs are separated ONLY by specia ...

I. Comparing genome sequences
... • Genomes become more dissimilar with greater phylogenetic distance ...
... • Genomes become more dissimilar with greater phylogenetic distance ...

BIOL2007 - EVOLUTIONARY TREES AND THEIR USES
... finding shortest trees compatible with the data. Aim is to assess of reliability of shortest tree compared to other trees that are almost as short (“near-parsimonious” trees). May use “bootstrapping”: run repeated phylogenetic analyses with random subsamples of the character data to test whether the ...
... finding shortest trees compatible with the data. Aim is to assess of reliability of shortest tree compared to other trees that are almost as short (“near-parsimonious” trees). May use “bootstrapping”: run repeated phylogenetic analyses with random subsamples of the character data to test whether the ...
