Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Oxidative phosphorylation wikipedia , lookup
Pharmacometabolomics wikipedia , lookup
Genetic code wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Biosynthesis wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Lactate dehydrogenase wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Citric acid cycle wikipedia , lookup
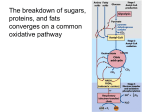
A 54-year-old female presents to her family physician's office with a 2 week history of pain and numbness in her left hand A 7-year-old boy is brought to the physician with a recent history of decreased activity A 63-year-old woman is brought to the physician for evaluation of her “parkinsonism” When do you suspect problems in metabolic pathways? Eric Niederhoffer Medical Biochemistry Metabolism in Skeletal Muscle and Nervous Tissue • Metabolism in skeletal muscle • Pathways overview • Regulation in skeletal muscle • Ischemic forearm test • Metabolism in nervous tissue • Pathways overview • Clinical aspects (muscle) • Clinical aspects (nervous tissue) • Clinical/laboratory findings • Examples of inherited metabolic disorders • Glycogen storage disease type VII • Pyruvate dehydrogenase complex deficiency • Maple syrup urine disease • Inborn errors of metabolism • Review questions Metabolism in Skeletal Muscle •Glycolysis •Glycogenolysis •-oxidation (ketone bodies) •Krebs (tricarboxylic acid) cycle •Branched-chain amino acids •Electron transport chain •Calcium regulation •Key enzyme regulation Pathways Overview Ketone bodies Glucose Glycogen Glycolysis Glycogenolysis Ca2+ G6P Fatty acids β-Oxidation Phosphorylase kinase a Lactate No O2 Branched-chain amino acids Ile, Leu, Val Ca2+ PDH Pyruvate Acetyl-CoA Electron Transport Chain Krebs cycle Ca2+ ICDH, αKGDH Production of ATP Regulation in Skeletal Muscle Ep Glc AR Glycolysis ATP Citrate AC cAMP PKA ATP G6P Ca2+ Phosphorylase kinase a F6P PFK-2 PP Pi IMP AMP PFK-1 F26BP NH4+ AMP Pi F16BP Glycogen Glycogenolysis PEP PDHP Ca2+ PDHP β-Oxidation Fatty acids PK PDHK PDH Pyr PDH Acetyl-CoA Ischemic Forearm Test • Obtain baseline lactate and ammonia levels • Inflate blood pressure cuff and perform repetitive rapid grip exercise • Once fatigued, remove cuff and obtain blood samples for lactate and ammonia levels • Normal result is elevated lactate and ammonia then return to baseline in 10-15 minutes Glucose Glycogen Glycolysis Glycogenolysis G6P hypoxia Lactate Pyruvate Acetyl-CoA Adenylate kinase 2ADP ATP NH4+ 2ATP AMP IMP AMP deaminase (hypoxia, low energy charge) Metabolism in Nervous Tissue •Glycolysis •Glycogenolysis (stress) •-oxidation (ketone bodies) •Krebs (tricarboxylic acid) cycle •Branched-chain amino acids •Electron transport chain Pathways Overview Glucose Glycogen Fatty acids Glycolysis Glycogenolysis Ketone bodies β-oxidation G6P Lactate (glia) Lactate No O2 Pyruvate Acetyl-CoA NH4+ + Glutamate (astrocytes) Gln synthetase Krebs cycle Glutamine Branched-chain amino acids Ile, Leu, Val Production of ATP Electron Transport Chain Clinical Aspects for Inborn Errors of Metabolism in Muscles Toxic accumulation disorders •Protein metabolism disorders (amino acidopathies, organic acidopathies, urea cycle defects) •Carbohydrate/intolerance disorders •Lysosomal storage disorders Energy production/utilization disorders •Fatty acid oxidation defects •Carbohydrate utilization, production disorders (glycogen storage, gluconeogenesis, and glycogenolysis disorders) •Mitochondrial disorders •Peroxisomal disorders •Metabolic acidosis (elevated anion gap) •Hypoglycemia •Hyperammonemia Clinical Aspects for Inborn Errors of Metabolism in Nervous Tissue Evidence of familial coincidence Progressive decline in nervous functioning Appearance and progression of unmistakable neurologic signs General symptoms •State of consciousness, awareness, reaction to stimuli •Tone of limbs, trunk (postural mechanisms) •Certain motor automatisms •Myotatic and cutaneous reflexes •Spontaneous ocular movements, fixation, pursuit; visual function •Respiration and circulation •Appetite •Seizures Clinical/Laboratory Findings Clinical findings AA OA UCD CD GSD FAD LSD PD MD Episodic decompensation X + ++ + X + - - X Poor feeding, vomiting, failure to thrive X + ++ + X X + + + Dysmorphic features and/or skeletal or organ malformations X X - - X X + X X Abnormal hair and/or dermatitis - X X - - - - - - Cardiomegaly and/or arrhythmias - X - - X X + - X Hepatosplenomegaly and/or splenomegaly X + + + + + + X X Developmental delay +/neuroregression + + + X X X ++ + + Lethargy or coma X ++ ++ + X ++ - - X Seizures X X + X X X + + X Hypotonia or hypertonia + + + + X + X + X Ataxia - X + X - X X - - Abnormal odor X + X - - - - - - Primary metabolic acidosis X ++ + + X + - - X Primary respiratory alkalosis - - + - - - - - - Hyperammonemia X + ++ X - + - - X Hypoglycemia X X - + X + - - X Liver dysfunction X X X + X + X X X Reducing substances X - - + - - - - - Ketones A H A A L/A L A A H/A Laboratory Findings* http://emedicine.medscape.com/article/804757-workup Examples of Inherited Metabolic Disorders Pentose Phosphate Pathway Glucose Glycogen R5P G6P Glycogenolysis Glycogenesis nucleotides Glycolysis Tarui disease Glycogen Storage Disease Type VII F6P PFK F16BP PDH complex deficiency PDH Pyruvate Acetyl-CoA Branched-chain amino acids Ile, Leu, Val Branched-chain -keto acids KMV, KIC, KIV BCKADH Maple syrup urine disease Branched-chain ketoaciduria Krebs cycle Glycogen Storage Disease Type VII (Tarui Disease) Classic, infantile onset, Late onset Exercise intolerance, fatigue, myoglobinuria Phosphofructokinase • Tetramer of three subunits (M, L, P) • Muscle/heart/brain - M4; liver/kidneys - L4; erythrocytes - M4, L4, ML3, M2L2, M3L General symptoms of classic form • Muscle weakness, pronounced following exercise • Fixed limb weakness • Muscle contractures • Jaundice • Joint pain Laboratory studies • Increased serum creatine kinase levels • No increase in lactic acid levels after exercise • Bilirubin levels may increase • Increased reticulocyte count and reticulocyte distribution width • Myoglobinuria after exercise • Ischemic forearm test - no lactate increase with ammonia increase Pyruvate Dehydrogenase Complex Deficiency Neonatal, infantile, childhood onset Abnormal lactate buildup (mitochondrial disease) Pyruvate dehydrogenase complex • E1 - (thiamine dependent) and subunits, 2 2 tetramer • E2 - monomer (lipoate dependent) • E3 - dimer (riboflavin dependent) common to KGDH and BCαKDH • X protein - lipoate dependent • Pyruvate dehydrogenase phosphatase Nonspecific symptoms (especially with stress, illness, high carbohydrate intake) • Severe lethargy, poor feeding, tachypnea • Key feature is gray matter degeneration with foci of necrosis and capillary proliferation in the brainstem (Leigh syndrome) • Infants with less than 15% PDH activity generally die Developmental nonspecific signs • Mental delays • Psychomotor delays • Growth retardation Laboratory studies • High blood and cerebrospinal fluid lactate and pyruvate levels • Elevated serum and urine alanine levels • If E2 deficient, elevated serum AAs and hyperammonemia • If E3 deficient, elevated BCAA in serum, KG in serum and urine • Ischemic forearm test – lactate increases baseline Maple Syrup Urine Disease (Branched-Chain -Ketoaciduria) Classic (early) and late onset (5 clinical phenotypes; classic, intermediate, intermittent, thiamine-responsive, and E3-deficient) Encephalopathy and progressive neurodegeneration Branched-chain -ketoacid dehydrogenase complex • E1 - (thiamine dependent) and subunits, 2 2 tetramer • E2 - monomer (lipoate dependent) • E3 - dimer (riboflavin dependent) common to KGDH and PDH • BCαKADH kinase • BCαKADH phosphatase Initial symptoms • Poor feeding, vomiting, poor weight gain, and increasing lethargy Neurological signs • Alternating muscular hypotonia and hypertonia, dystonia, seizures, encephalopathy Laboratory studies • Elevated BCAA in serum • Presence of alloisoleucine in serum • Presence of -HIV, lactate, pyruvate, and KG in urine Treatment • Restriction of BCAA • Supplementation with thiamine Inborn Errors of Metabolism Carbohydrates (Glycogen storage diseases) Amino acids (Maple syrup urine disease) Organic acids (Alkaptonuria) Mitochondrial function (Pyruvate dehydrogenase deficiency) Purines and pyrimidines (Lesch-Nyhan disease) Lipids (Familial hypercholesterolemia) Porphyrins (Crigler-Najjar syndromes) Metals (Hereditary hemochromatosis) Peroxisomes (X-linked adrenoleukodystrophy) Lysosomes (GM2 gangliosidoses - Tay Sachs disease) Hormones (hyperthyroidism) Blood (Sickle cell disease) Connective tissue (Marfan syndrome) Kidney (Alport syndrome) Lung (1-antitrypsin deficiency) Skin (Albinism) For additional examples (Year Two Review 1, 2) Review Questions • How does muscle produce ATP (carbohydrates, fatty acids, ketone bodies, branched-chain amino acids)? • How is skeletal muscle phosphofructokinase-1 regulated? • What are the key Ca2+ regulated steps? • How does nervous tissue (neurons and glial cells) produce ATP (carbohydrates, fatty acids, ketone bodies, branched-chain amino acids)? • How do glial cells (astrocytes) assist neurons? • What are some key clinical features (history, physical, laboratory test results) associated with defects in metabolism that affect muscles and nervous tissue?



















![fermentation[1].](http://s1.studyres.com/store/data/008290469_1-3a25eae6a4ca657233c4e21cf2e1a1bb-150x150.png)