Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
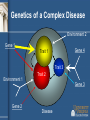
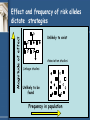
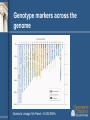
Genetic Epidemiology Michèle Sale, Ph.D. Center for Public Health Genomics [email protected] Tel: 982-0368 Genetic epidemiology • “A science which deals with the etiology, distribution, and control of disease in groups of relatives and with inherited causes of disease in populations.” Newton E. Morton, 1982 Model for Complex Diseases + = Disease Susceptibility Genetics of a Complex Disease Environment 2 Gene 1 Gene 4 Trait 1 Trait 3 Environment 1 Gene 2 Trait 2 Gene 3 Disease “Monogenic” vs “Complex” Disease Mendelian Complex 1 or small # of genes Many Often etiologic (severe phenotype) Susceptibility / molecular pathology ? Highly penetrant Modest penetrance High Odds Ratio Modest/Low Odds Ratio Strong selection => Low frequency/Rare Weak/No selection => High frequency/Common Coding Sequence Non-coding/regulation (?) Overall steps for disease gene identification • • • • • Is there a genetic component? Study design Measurement of phenotype Molecular analysis Functional analysis Is there a genetic component? • • • • Twin studies Familial aggregation Segregation analysis Race/ethnicity differences Twin studies • • • • • Comparison of monozygotic (MZ) pairs (who share all their genome) with dizygotic (DZ) twins (who share half of their genome in common on average, same as sibs) The greater similarity of MZ twins than DZ twins is considered evidence of genetic factors The pairwise (Pr) concordance (Pr) is the proportion of affected pairs that are concordant for the disease. – The proportion of twin pairs with both twins affected of all ascertained twin pairs with at least one affected – Pr=C/(C+D), where C is the number of concordant pairs and D is the number of discordant pairs. The probandwise (Cc) concordance is the proportion of affected individuals among the co-twins of previously ascertained index cases. – Allows for double counting of doubly ascertained twin pairs and is interpretable as the recurrence risk in a co-twin of an affected individual – Cc=2C/(2C+D) In theory, complete genetic determination of a disease would equate to MZ twins having 100% concordance and DZ twins having 50% concordance Twin studies - assumptions • Random mating • No interactions between genes and environment • Equivalent environments for MZ and DZ twins Concordance rates for some traits Trait Blood pressure Pulse rate Measles Diabetes mellitus Tuberculosis Schizophrenia Manic-depressive psychosis MZ 63% 56% 95% 85% 54% 58% 76% DZ 36% 34% 87% 37% 22% 10% 18% Other types of twin studies • Twins discordant for disease have been used to examine possible environmental causes. • Adoption studies also permit the separation of childhood rearing effects from genetic effects by studying the similarity of adopted children with their biological and adoptive parents Bouchard et al. Science. 1990 Oct 12;250(4978):223-8. Familial aggregation • Sibling risk relative ratio ls = risk to a sibling of person with disease of interest population risk SNP Disease Variant & λs in Diabetes Type 1 Type 2 Prob of Disease (Sibling) 6% 30-40% Prob of Disease (Unrelated) 0.4% 7% λs 15 4-6 Recurrence risks for multiple sclerosis in families Compston and Coles. Lancet. 2002;359(9313):1221-31. Segregation analysis • Determines which specific model (genetic or environmental) best fits the familial aggregation • E.g. – Major gene or many genes (polygenic)? – Dominant, additive, recessive inheritance? Differences in prevalence across race/ethnicities Time trends in the percentage of African American adolescents and adults who were overweight, 1988-94 http://www.niddk.nih.gov/health/diabetes/pubs/afam/afam.htm Adiposity • However, rates of type 2 diabetes in African Americans still higher than Caucasian Americans after controlling for age, adiposity, and socio-economic status • Other factors must be involved Epidemiological study designs http://en.wikipedia.org/wiki/Study_design Study designs • Case series: what clinicians see • Case-control: compare people with and without a disease • Cohort: follow people over time to see who gets the disease • Randomized controlled trial (RCT) Other terms • Retrospective vs. prospective • Cross-sectional vs. longitudinal Measurement of Phenotype • What is the phenotype? – e.g., diabetes, fasting glucose, oral glucose tolerance test • How is it diagnosed? – Physician’s diagnosis, clinical measurements, questionnaire • How objective are the phenotypes? – Physician’s diagnosis – somewhat variable – Clinical measurements – most pretty good *The more defined the phenotype, the easier to find the gene(s) that controls it Additional consideration in genetic studies: families or unrelated individuals? Patient ascertainment • Sib-pair: • Families: • Case-control: ? ? ? or ? Molecular/Analytical approaches • Linkage – Families • Association – – – – Candidate gene Genome wide Generally case-control There are family-based approaches Magnitude of effect Effect and frequency of risk alleles dictate strategies Unlikely to exist Association studies Linkage studies Unlikely to be found Frequency in population Linkage analysis • Linkage = The proximity of two or more markers on a chromosome • Linkage analysis is a statistical method for detecting linkage between a disease and markers of known location by following their inheritance in families • Uses recombination to define genomic interval likely to contain gene/s • Single large pedigree or multiple small pedigrees Linkage analysis • • • • • • • Works well for Mendelian traits and more highly penetrant diseases Low resolution = fewer markers needed and resilient to allelic heterogeneity Apparently high Type I error rate for complex/non-Mendelian diseases – more loci, common variants, high phenocopy rate, and lower penetrance Large pedigrees better for rare alleles – more likely to segregate the allele Large pedigrees increase the probability of parental heterozygosity for frequent alleles Most likely to detect intermediate frequency alleles Strong pedigree signal may reflect rare Mendelian forms of complex disease – eg BRCA1 & BRCA2 mutations in breast and ovarian cancer Genotype markers across the genome Illumina's Linkage IVb Panel: >6,000 SNPs LOD score • LOD score = Log of the Odds of linkage = log10 Likelihood of linkage Likelihood of not being linked = log10 L(Q<0.5) L(Q=0.5) • The closer two markers are to each other, the lower the odds of a recombination (crossing over event) occurring between them in meiosis. Linkage analysis Is there cosegregation of a chromosomal region with the phenotype? Linkage analysis Is there cosegregation of a chromosomal region with the phenotype? Linkage analysis Is there cosegregation of a chromosomal region with the phenotype? • Add additional markers to region • Add additional families to study Association study • Best power for common variants of modest - low effect size • Search for specific genetic differences distinguishing cases from controls Cases Controls Case - Control is the most popular study design for complex disease genetics: • Cross-sectional - no follow-up • More efficient recruitment than families - easy to ascertain and recruit • Easy to analyze • Statistical power compared to family-based linkage X • Cases and controls must be wellmatched - Drawn from same population - Randomize non-genetic confounder factors • At risk for type 1 errors if incomplete matching (stratification) Genome-wide association studies (GWAS): A paradigm shift in human genetics How can we use SNPs to find diabetes genes? • Genome-Wide Association Study (GWAS) – Examination of variation across the entire human genome to identify genetic correlations with the presence or absence of diabetes • Two groups: cases (have diabetes) vs. controls (don’t have diabetes) • Each participant’s genome is surveyed for markers of genetic variation (SNPs) • Groups compared to determine specific genetic differences between the two groups GWAS approach • • • http://www.genizon.com/html/gestion/Research_Technology.jpg Does not assume knowledge of genes/biology Investigate markers evenly spaced along genome Investigate association: Joint occurrence of two alleles (e.g. disease allele and marker allele) in a population > expected frequency Why are GWAS now feasible? • SNP identification efforts more SNPs in databases • Understanding of linkage disequilibrium in the human genome (HapMap project) fewer “tagSNPs” to genotype • Lower cost of genotyping platforms Products now use >1 million SNPs! Pairwise tagging A/T 1 A A T T G/A 2 G G A A high r2 G/C 3 G C G C T/C 4 T C C C G/C 5 A/C 6 A C C C G C G C high r2 After Carlson et al. (2004) AJHG 74:106 http://www.hapmap.org/downloads/presentations/2_Daly.ppt high r2 Tags: SNP 1 SNP 3 SNP 6 3 in total Test for association: SNP 1 SNP 3 SNP 6 Use of haplotypes can improve genotyping efficiency A/T 1 G/A 2 G/C 3 T/C 4 G/C 5 A/C 6 Tags: SNP 1 SNP 3 A A T T G G A A G C G C T C C C G C G C http://www.hapmap.org/downloads/presentations/2_Daly.ppt A C C C 2 in total Test for association: SNP 1 captures 1+2 SNP 3 captures 3+5 “AG” haplotype captures SNP 4+6 Relative power (%) Efficiency and power tag SNPs random SNPs ~300,000 tag SNPs needed to cover common variation in whole genome in CEU Average marker density (per kb) P.I.W. de Bakker et al. (2005) Nat Genet Genotyping platform: Illumina Cost: 370 Duo $240-280 650Y $480-520 1M $580-650 http://www.genengnews.com/articles/chtitem.aspx?tid=1862&chid=2 Genotyping platform: Affymetrix http://gmed.bu.edu/about/genotyping.html YRI Completeness of dbSNP • CEU Vast majority of common SNPs are contained in or highly correlated with a SNP in dbSNP CHN+JPT Nature 437, 1299-1320. 2005 Comparison of coverage YRI Platform CEU CHB+JPT % r2 ≥ 0.8Mean max r2% r2 ≥ 0.8Mean max r2 % r2 ≥ 0.8Mean max r2 Affymetrix GeneChip 500K 46 0.66 68 0.81 67 0.80 Affymetrix SNP Array 6.0 66 0.80 82 0.90 81 0.89 Illumina HumanHap300 33 0.56 77 0.86 63 0.78 Illumina HumanHap550 55 0.73 88 0.92 83 0.89 Illumina HumanHap650Y 66 0.80 89 0.93 84 0.90 Perlegen 600K 47 0.68 92 0.94 84 0.90 Paul de Bakker, pers. comm. Association Studies • Detect genes/genomic regions associated with a disease through allelic associations in case-control studies – Causal variants are associated with disease phenotype – Linked neutral variants are associated with the disease phenotype through LD with the causal variant • Younger disease variants (rarer variant) – LD around the variant is stronger = better power – Associated region containing variant is broad (low genome resolution) • Older disease variants (common variant) – Weaker LD = worse power – Better association map resolution Phenotype-genotype association Marker associated with disease could be: 1. False positive result (type 1 error) 2. Co-inherited with a true causative (functional) variant 3. A true functional or causative variant Replication and follow-up • Many analytical tests – high probability of false positives • Replicate in additional studies (often requires crossstudy collaboration) • Map the causal variant – Denser marker map – Evidence for other variants in the same gene with (perhaps smaller) independent effects (allelic heterogeneity) – Haplotype analysis – Resequencing – Sequence / genome mapping bioinformatics to identify or predict genome features in the linkage disequilibrium region of the map SNP – Expression or reporter assays Common Disease Common Variant Hypothesis (CDCV) • Genetic risk for common diseases (diabetes, CHD, hypertension, schizophrenia, asthma,..) results from common variants/polymorphisms in multiple genes • The effects for each gene variant must be smaller than in monogenic disorders otherwise the prevalence of the diseases would be very high • Since SNPs are a common mode of variation in the human genome - and coding SNPs lead to mongenic diseases - SNPs may be the variants that are associated with risk for common diseases Do the Common Disease Variants Code ? Not necessarily (or usually ?) • Protein coding SNPs (cSNPs) may disrupt protein fold, structure, activity • Variants in Mendelian diseases with high penetrance (50-100% penetrance) often disrupt proteins • But common diseases are not penetrant to the same level - genetic odds ratios : Prob (Disease | risk allele) ~ 1.2 -1.5 Prob (Not Disease | risk allele) Early successes • Klein R et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005 Apr 15; 308:385-9. – 96 cases and 50 controls; 116,204 SNPs • Maraganore et al. High-resolution wholegenome association study of Parkinson disease. Am J Hum Genet. 2005 Nov; 77:685-93. – 198,345 SNPs in 443 sibling pairs discordant for PD – 1,793 PD-associated SNPs (P<.01 in tier 1) and 300 genomic control SNPs in 332 matched case-unrelated control pairs The future • Identify additional genes in diverse populations • Identify causal variant/s in these genes • Determine function of novel genes, and function of causative variants • Explore gene x gene interactions (epistasis) and gene x environment interactions (e.g. physical activity, diet) • Other technological advances: – – – – Animal models of disease Innovative imaging of target tissues Functional approaches to gene expression profiling Whole genome sequencing? • Era of “personalized medicine”&/or prevention END Questions?