Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
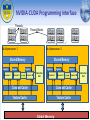
Accelerating HMMER Search on GPUs using Hybrid Task and Data Parallelism Narayan Ganesan1, Roger Chamberlain2, Jeremy Buhler2 and Michela Taufer1 Computer and Info. Sciences Dept, University of Delaware.1 Computer Science and Engineering, Washington University in St. Louis2 Motivation • Dataset size is growing for many applications rapidly • Problem sizes grow as the product of the data sizes • • • • • E.g., Genome sequence alignment, protein motif finding Number of problems also grows as the product of number of available data Many sequentially dependent algorithms are executed serially Processor speed has hit a brick wall (~3.5-4.00GHz in 2003) and serial evaluation is just not feasible for large data applications Need for parallelism is greater than ever Parallel hardware is ubiquitous GPU, Multi-core, SIMD, Hybrid, MIMD, FPGA Efforts must be spent on parallelizing algorithms and applications for these hardware Application: Protein Motif Finding • Proteins are synthesized by biological Processes that can be described by HMMs Class A Protein Synthesis . . . Class N Protein Synthesis ...PAQVEMYKFLLRISQLNRD... ... CHTEARGLEGVCIDPKK... ... DGEACNSPYLDWRKDTEQ... ... CTPPSLAACTPPTS... ... LTITNLMKSLGFKPKPKKI... ... DELAAMVRDYLKKTPEF... • Each class is described by short characteristic sequences or motifs • Each class or “generator” is described by a Hidden Markov model • Protein Motif Finding answers the questions: Given a sequence what class does it belong to? Given a sequence and a HMM what is the probability that the sequence belongs to that class? Protein Motif Finding • Protein motif finding is very similar to signal source identification • A protein sequence may contain multiple motifs: ..ACGFTDFWAPSLTHLTIKNL.. • Motifs in the sequence are sometimes modified by addition and deletion • • • of random amino acids Occurrence of motifs can be modeled by Profile Hidden Markov models Viterbi algorithm is used to “decode” a given protein sequence against a model The result is the probability that the sequence belongs to the class Protein Synthesis ...PAQVEMYKFLLRISQLNRD... ... CHTEARGLEGVCIDPKK... ... DGEACNSPYLDWRKDTEQ... HMMER Search - Protein Motif Finding Sample Paths S N B D4 D2 D3 I1 I2 I3 I4 M1 M2 M3 M4 M5 E C T J ....ACGFTDFWAPSLTHLTIKNL.... ....ACGFTDFWAPSLTHLTIKNL.... ....ACGFTDFWAPSALTHLTIKNL.... ....ACGFTDFWAPSAGLTHLTIKNL... ....ACGFTDFWAPSAGL-HLTIKNL... HMMER Search – Recurrence Equations D2 S N B D3 D4 I1 I2 I3 I4 M1 M2 M3 M4 J VD VI VM M5 E C T HMMER Search- Protein Motif Finding 1 1 Model j m Insert: S e q u e n c e XE Match: Delete: i VD (i, j ) max( VD (i, j 1) ci , di ) L • Dependence on XE imposes a row major order computation • Delete state costs impose a serial dependency on previous element in the row • Harder to parallelize by conventional means HMMER Search – Protein Motif Finding • Delete costs impose sequential dependency VD (i,3k ) max( VD (i,2k ) c3k , d3k ) VD (i, k ) max( VD (i,1) ck , d k ) 1 m row i VD (i,2k ) max( VD (i, k ) c2 k , d 2 k ) VD (i, m) max( VD (i,3k ) cm , d m ) • Parallelize the row calculations • The recurrence for VD is parallelizable by blocking strategy NVIDIA-CUDA Programming Interface Threads Thread Blocks ... ... Multiprocessor 1 Multiprocessor 2 Shared Memory Registers Processor 1 Registers Processor 2 … Shared Memory Registers Processor M Registers Instruction Unit Processor 1 Registers Processor 2 … Registers Processor M Constant Cache Constant Cache Texture Cache Texture Cache Global Memory Instruction Unit Traditional Task Parallelism for GPUs • Different GPU threads work on independent tasks • Time taken for different tasks vary according to the nature of the job Different sequences of different lengths take different time to complete Load imbalance issues are possible Total time depends on location of the longest sequences in the database Seq i Thread Block 1 Seq i+M Thread Block 2 Thread Block P Hybrid data and task parallelism • We extract parallelism out of data • • dependency Multiple GPU threads cooperate to work on the same task By dividing the database into roughly equal chunks, we naturally solve any load imbalance problems This technique works for uniform recurrence equations Ubiquitous in computational biology including local sequence alignment, multiple sequence alignment, motif finding Seq i Thread Block 1 Seq i+M Thread Block 2 Thread Block P GPU Implementation row i 1 m • Multiple threads cooperate by partitioning a single sequence and • • • working on different partitions Working set is one row of the DP matrix, 507x3, 32-bit integers Model of size 507 has 507x9 transition probabilities and 507x40 emission probabilities stored as short integers Model data is read in a coalesced form into the shared memory Working set is stored and updated within shared memory Performance and Results • We compared our implementation on 1 and 4 GPUs versus the mpi-gpu HMMER for the same dataset 3 HMM Sizes: • 128 • 256 • 507 NCBI NR Protein Database: • 5.5GB in Size • 10.5 Million Protein Sequences Conclusions and Future Work • Our GPU implementation of HMMER is: 5-8x faster than GPU-HMMER search implementation for the same data set 100x faster than the CPU implementation • Future work: Phylogenetic Motif Identification Identify common genetic motifs among several groups of organisms Representative of evolutionary relatedness among different (typically 1000s) species Closely related to multiple sequence alignment problem via Hidden Markov Models Computationally intensive for which faster motif finding is absolutely necessary