Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Site-specific recombinase technology wikipedia , lookup
Protein moonlighting wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome evolution wikipedia , lookup
Public health genomics wikipedia , lookup
Helitron (biology) wikipedia , lookup
Pharmacogenomics wikipedia , lookup
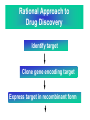
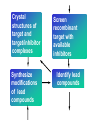
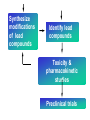
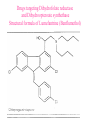
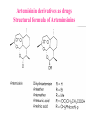
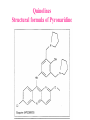
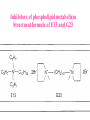
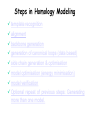
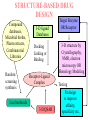
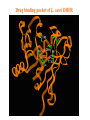
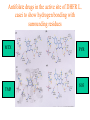
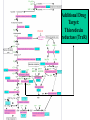
Case Study #1 Use of bioinformatics in drug development and diagnostics Ashok Kolaskar University of Pune, Pune 411 007, India. [email protected] Bringing a New Drug to Market Review and approval by Food & Drug Administration 1 compound approved Phase III: Confirms effectiveness and monitors adverse reactions from long-term use in 1,000 to 5,000 patient volunteers. Phase II: Assesses effectiveness and looks for side effects in 100 to 500 patient volunteers. 5 compounds enter clinical trials Phase I: Evaluates safety and dosage in 20 to 100 healthy human volunteers. 5,000 compounds evaluated 0 2 4 6 8 Discovery and preclininal testing: Compounds are identified and evaluated in laboratory and animal studies for safety, biological activity, and formulation. 10 Source: Tufts Center for the Study of Drug Development 12 14 Years 16 Biological Research in 21st Century “ The new paradigm, now emerging is that all the 'genes' will be known (in the sense of being resident in databases available electronically), and that the starting "point of a biological investigation will be theoretical.” - Walter Gilbert Rational Approach to Drug Discovery Identify target Clone gene encoding target Express target in recombinant form Crystal structures of target and target/inhibitor complexes Synthesize modifications of lead compounds Screen recombinant target with available inhibitors Identify lead compounds Synthesize modifications of lead compounds Identify lead compounds Toxicity & pharmacokinetic studies Preclinical trials Case study: Malaria • P. falciparum presents a fascinating model system. The life-cycle complexity is both a challenge and an opportunity. • Unfortunately at the molecular level much remains unknown. • Its genome, currently being sequenced, is already yielding valuable data. Malarial Drugs • Up until now, nearly all prophylaxis and therapeutic intervention has been based on traditional medicines or their derivatives eg quinine, paraquine, chloroquine etc. • Effective also have been type I antifolates eg sulphones and sulphonamides which mimic PABA, and the type II antifolates eg pyrimethamine, trimethoprim and proguanil which mimic dihydrofolate. List of Drugs • Chloroquine – Avloclor ®; Nivaquine® • Antifolate – Pyrimethamine (Daraprim®) – Proguanil (Paludrine®) • Combination drugs – Pyrimethamine & Sulfadoxine combination eg.: Fansidar ® Resistance to these drugs poses a threat to control morbidity and mortality of malaria. Drug Resistance (DR) • Mechanisms of drug resistance – Formation of altered target • Target has decreased affinity for substrate/analogue – Decreased access to target • Mutations decrease membrane permeability • Specific transport systems altered/deleted – Increased level of enzymes which cleave substrates • e.g. Increased expression of -lactamase • Gene amplification – Decreased activation of drug • Drugs require activation by enzymes near site • Activation pathway suppressed/deleted Malaria: Novel drug combinations Structural formula: Atovaquone & Proguanil Drugs targeting Dihydrofolate reductase and Dihydroopteroate synthethase Structural formula of Lumefantrine (Benflumethol) Artemisinin derivatives as drugs Structural formula of Arteminsinins Quinolines Structural formula of Pyronaridine Novel drug combinations Structural formula of Chlorproguanil and Dapsone Dihydrofolate reductase inhibitors Structural formula of Pyrimethamine & Cycloguanil Inhibitors of phospholipid metabolism Structural formula of E13 and G23 An Ideal Target • Is generally an enzyme/receptor in a pathway and its inhibition leads to either killing a pathogenic organism (Malarial Parasite) or to modify some aspects of metabolism of body that is functioning dormally. • An ideal target… – – – – – Is essential for the survival of the organism. Located at a critical step in the metabolic pathway. Makes the organism vulnerable. Concentration of target gene product is low. The enzyme amenable for simple HTS assays How Bioinformatics can help in Target Identification? • • • • • Homologous & Orthologous genes Gene Order Gene Clusters Molecular Pathways & Wire diagrams Gene Ontology Identification of Unique Genes of Parasite as potential drug target. Comparative Genomics of Malarial Parasites: Source for identification of new target molecules. • Genome comparisons of malarial parasites of human. • Genome comparisons of malarial parasites of human and rodent. • Comparison of genomes of – – Human – Malarial parasite – Mosquito What one should look for? Human P.f Mosquito Proteins that are shared by – •All genomes •Exclusively by Human & P.f. •Exclusively by Human & Mosquito •Exclusively by P.f. & Mosquito Unique proteins in – Human P.f. Targets for anti-malarial drugs Mosquito What is Structural Genomics? • Organise all known proteins into families. • Determine structures of at least one member of every family. • Solve structures of more than 10,000 protein in next 10 years. • Generate knowledge and rules from known protein structures. • Apply this knowledge to predict the structure of each and every protein of known organisms. Objectives of Structural Genomics • Selection of Targets for structure determination to obtain maximum information return on total efforts • Develop mechanism that facilitates cooperation and prevent work duplication Impact of Structural Genomics on Drug Discovery Dry, S. et. al. (2000) Nat. Struc.Biol. 7:976-949. Drug Development Flowchart • Check if structure is known • If unknown, model it using KNOWLEDGE-BASED HOMOLOGY MODELING APPROACH. • Search for small molecules/ inhibitors • Structure-based Drug Design • Drug-Protein Interactions • Docking Why Modeling? • Experimental determination of structure is still a time consuming and expensive process. • Number of known sequences are more than number of known structures. • Structure information is essential in understanding function. Sequence identities & Molecular Modeling methods Methods Sequence Identity with known structures • ab initio 0-20% • Fold recognition 20-35% • Homology Modeling >35% What is Homology or Comparative Modeling? • Comparisons of the tertiary structures of homologous proteins have shown that 3-D structures have been better conserved during evolution than protein primary structures. • In the absence of experimental data, modelbuilding on the basis of the known three dimensional structure of homologous protein(s) is the only reliable method to obtain structural information. Difference between Homology and Similarity • Homology does not necessarily imply similarity. • Homology has a precise definition: having a common evolutionary origin. • Since homology is a qualitative description of the relationship, the term “% homology” has no meaning. • Supporting data for a homologous relationship may include sequence or structural similarities, which can be described in quantitative terms. Sequence Identity Model Accuracy Rate limiting factors in modeling 100% CPU time to model 75% Quality of model & Loop modeling 50% Errors in the sequence alignment 25% Detection of homology What is Remote Homology Modeling? • Modeling based on low levels of sequence identity (<30%). • Has 3 obstacles to overcome: the remote homology has to be detected; Q and T have to be aligned correctly; homology modeling procedure has to be tailored to the harder problem of extremely low sequence identity. Steps in Homology Modeling template recognition alignment backbone generation generation of canonical loops (data based) side chain generation & optimisation model optimisation (energy minimisation) model verification Optional repeat of previous steps: Generating more than one model. STRUCTURE-BASED DRUG DESIGN Compound databases, Microbial broths, Plants extracts, Combinatorial Libraries Random screening synthesis 3-D ligand Databases Docking Linking or Binding Receptor-Ligand Complex Lead molecule 3-D QSAR Target Enzyme OR Receptor 3-D structure by Crystallography, NMR, electron microscopy OR Homology Modeling Testing Redesign to improve affinity, specificity etc. Binding Site Analysis • In the absence of a structure of Targetligand complex, it is not a trivial exercise to locate the binding site!!! • This is followed by Lead optimization. Lead Optimization Active site Lead Lead Optimization Factors Affecting The Affinity Of A Small Molecule For A Target Protein LIGAND.wat n +PROTEIN.wat n LIGAND.PROTEIN.watp+(n+m-p) wat • HYDROGEN BONDING • HYDROPHOBIC EFFECT • ELECTROSTATIC INTERACTIONS • VAN DER WAALS INTERACTIONS • STRAIN IN THE LIGAND ( BOUND) • STRAIN IN THE PROTEIN DIFFERENCE BETWEEN AN INHIBITOR AND DRUG Extra requirement of a drug compared to an inhibitor •Selectivity LIPINSKI’S RULE OF FIVE Poor absorption or permeation are more •Less Toxicity likely when : •Bioavailability -There are more than five H-bond donors •Slow Clearance -The mol.wt is over 500 Da •Reach The Target -The MlogP is over 4.15(or CLOG P>5) •Ease Of Synthesis -The sums of N’s and O’s is over 10 •Low Price •Slow Or No Development Of Resistance •Stability Upon Storage As Tablet Or Solution •Pharmacokinetic Parameters •No Allergies THERMODYNAMICS OF RECEPTOR-LIGAND BINDING •Proteins that interact with drugs are typically enzymes or receptors. •Drug may be classified as: substrates/inhibitors (for enzymes) agonists/antagonists (for receptors) •Ligands for receptors normally bind via a non-covalent reversible binding. •Enzyme inhibitors have a wide range of modes:non-covalent reversible,covalent reversible/irreversible or suicide inhibition. •Enzymes prefer to bind transition states (reaction intermediates) and may not optimally bind substrates as part of energy used for catalysis. •In contrast, inhibitors are designed to bind with higher affinity: their affi nities often exceed the corresponding substrate affinities by several orders of magnitude! •Agonists are analogous to enzyme substrates: part of the binding energy may be used for signal transduction, inducing a conformation or aggregation shift. •To understand ‘what forces’ are responsible for ligands binding to Receptors/Enzymes, •It is worthwhile considering what forces drive protein folding –they share many common features. •The observed structure of Protein is generally a consequence of the hydrophobic effect! •Secondary amides form much stronger H-bonds to water than to other sec. Amides hydrophobic collapse •Proteins generally bury hydrophobic residues inside the core,while exposing hydrophilic residues to the exterior Salt-bridges inside •Ligand building clefts in proteins often expose hydrophobic residues to solvent and may contain partially desolvated hydrophilic groups that are not paired: •The desolvation penalty is paid for by favourable (hydrophobic) interaction elsewhere in the structure. Docking Methods • Docking of ligands to proteins is a formidable problem since it entails optimization of the 6 positional degrees of freedom. • Rigid vs Flexible • Speed vs Reliability • Manual Interactive Docking GRID Based Docking Methods • Grid Based methods – GRID (Goodford, 1985, J. Med. Chem. 28:849) – GREEN (Tomioka & Itai, 1994, J. Comp. Aided. Mol. Des. 8:347) – MCSS (Mirankar & Karplus, 1991, Proteins, 11:29). • Functional groups are placed at regularly spaced (0.3-0.5A) lattice points in the active site and their interaction energies are evaluated. Automated Docking Methods • Basic Idea is to fill the active site of the Target protein with a set of spheres. • Match the centre of these spheres as good as possible with the atoms in the database of small molecules with known 3-D structures. • Examples: – DOCK, CAVEAT, AUTODOCK, LEGEND, ADAM, LINKOR, LUDI. Folate Biosynthetic pathway DHFR CLUSTAL W (1.81) multiple sequence alignment chabaudi vinckei berghei yoelii vivax falciparum -----------------------E--KAGCFSNKTFKGLGNEGGLPWKCNSVDMKHFSSV -----------AICACCKVLNSNE--KASCFSNKTFKGLGNAGGLPWKCNSVDMKHFVSV MEDLSETFDIYAICACCKVLNDDE--KVRCFNNKTFKGIGNAGVLPWKCNLIDMKYFSSV -----------AICACCKVINNNE--KSGSFNNKTFNGLGNAGMLPWKYNLVDMNYFSSV MEDLSDVFDIYAICACCKVAPTSEGTKNEPFSPRTFRGLGNKGTLPWKCNSVDMKYFSSV -------------------------KKNEVFNNYTFRGLGNKGVLPWKCNSLDMKYFCAV * *. **.*:** * **** * :**::* :* 35 47 58 47 60 35 chabaudi vinckei berghei yoelii vivax falciparum TSYVNETNYMRLKWKRDRYMEK---------NNVKLNTDGIPSVDKLQNIVVMGKASWES TSYVNENNYIRLKWKRDKYIKE---------NNVKVNTDGIPSIDKLQNIVVMGKTSWES TSYINENNYIRLKWKRDKYMEKHNLK-----NNVELNTNIISSTNNLQNIVVMGKKSWES TSYVNENNYIRLQWKRDKYMGKNNLK-----NNAELNNGELN--NNLQNVVVMGKRNWDS TTYVDESKYEKLKWKRERYLRMEASQGGGDNTSGGDNTHGGDNADKLQNVVVMGRSSWES TTYVNESKYEKLKYKRCKYLNKET----------VDNVNDMPNSKKLQNVVVMGRTNWES *:*::*.:* :*::** :*: * .:***:****: .*:* 86 98 113 100 120 85 chabaudi vinckei berghei yoelii vivax falciparum IPSKFKPLQNRINIILSRTLKKEDLAKEYN------NVIIINSVDDLFPILKCIKYYKCF IPSKFKPLENRINIILSRTLKKENLAKEYS------NVIIIKSVDELFPILKCIKYYKCF IPKKFKPLQNRINIILSRTLKKEDIVNENN--NENNNVIIIKSVDDLFPILKCTKYYKCF IPPKFKPLQNRINIILSRTLKKEDIANEDNKNNENGTVMIIKSVDDLFPILKAIKYYKCF IPKQYKPLPNRINVVLSKTLTKEDVK---------EKVFIIDSIDDLLLLLKKLKYYKCF IPKKFKPLSNRINVILSRTLKKEDFD---------EDVYIINKVEDLIVLLGKLNYYKCF ** ::*** ****::**:**.**:. * **..:::*: :* :***** 140 152 171 160 171 136 chabaudi vinckei berghei yoelii vivax falciparum I----------------------------------------------------------IIGGASVYKEFLDRNLIKKIYFTRINNAYT-----------------------------IIGGSSVYKEFLDRNLIKKIYFTRINNSYNCDVLFPEINENLFKITSISDVYYSNNTTLD IIGGSYVYKEFLDRNLIKKIYFTRINNSYN-----------------------------IIGGAQVYRECLSRNLIKQIYFTRINGAYPCDVFFPEFDESQFRVTSVSEVYNSKGTTLD I----------------------------------------------------------* 141 182 231 190 231 137 chabaudi vinckei berghei yoelii vivax falciparum ----------------FIIYSKTKE 240 --------FLVYSKVGG 240 --------- Multiple alignment of DHFR of Plasmodium species Drug binding pocket of L. casei DHFR Antifolate drugs in the active site of DHFR L. casei to show hydrogen bonding with surrounding residues MTX TMP PYR SO3 Prediction & Design of New Drugs • Prediction of 3-D PfDHFR using bacterial DHFR and homology modeling approach. • Search for the compounds using bifunctional basic groups that could form stable H-bonds in a plane with carboxyl group. • Optimize the structure of small molecules and then dock them on PfDHFR model. • Toyoda et. al. (1997). BBRC 235:515-519 could identify two compounds. How molecular modeling could be used in identifying new leads • These two compounds a triazinobenzimidazole & a pyridoindole were found to be active with high Ki against recombinant wild type DHFR. • Thus demonstrate use of molecular modeling in malarial drug design. Additional Drug Target: glutathione-GR Glutathione-GR Additional Drug Target: Thioredoxin reductase (TrxR) Cancer cell growth appears to be related to evolutionary development of plump fruits and vegetables • Large tomatoes can evolve from wild, blueberry-size tomatoes. The genetic mechanism responsible for this is similar to the one that proliferates cancer cells in mammalians. • This is a case where we found a connection between agricultural research, in how plants make edible fruit and how humans become susceptible to cancer. That's a connection nobody could have made in the past. Cornell University News, July 2000 Size of Tomato Fruit Single gene, ORFX, that is responsible for QTL has a sequence and structural similarity to the human oncogene c-H-ras p21. Fruit size alterations, imparted by fw2.2 alleles, are most likely to be due to the changes in regulation rather than in sequence/structure of protein. •fw2.2: A Quantitative Trait Locus (QTL) key to the Evolution of Tomato Fruit Size. Anne Frary (2000) Science, 289: 85-88 Genome Update: Public domain • Published Complete Genomes: 59 – Archaeal 9 – Bacterial 36 – Eukaryal 14 • Ongoing Genomes: 335 – Prokaryotic 203 – Eukaryotic 132 Private sector holds data of more than 100 finished & unfinished genomes. Challenges in Post-Genomic era: Unlocking Secretes of quantitative variation • For even after genomes have been sequenced and the functions of most genes revealed, we will have no better understanding of the naturally occurring variation that determines why one person is more disease prone than another, or why one variety of tomato yields more fruit than the next. • Identifying genes like fw2.2 is a critical first step toward attaining this understanding. Value of Genome Sequence Data • Genome sequence data provides, in a rapid and cost effective manner, the primary information used by each organism to carry on all of its life functions. • This data set constitutes a stable, primary resource for both basic and applied research. • This resource is the essential link required to efficiently utilize the vast amounts of potentially applicable data and expertise available in other segments of the biomedical research community. Challenges • Genome databases have individual genes with relatively limited functional annotation (enzymatic reaction, structural role) • Molecular reactions need to be placed in the context of higher level cellular functions The “omics” Series • Genomics – Gene identification & charaterisation • Transcriptomics – Expression profiles of mRNA • Proteomics – functions & interactions of proteins • Structural Genomics – Large scale structure determination • Cellinomics – Metabolic Pathways – Cell-cell interactions • Pharmacogenomics – Genome-based drug design Different levels of function Typically present in genome database • Atomic: Binds ATP • Molecular reaction: adds phosphate (phosphofructokinase) • Pathway: Gluconeogenesis • Network: Energy metabolism Typically, little informatics support for making these connections Data Mining: Finding the Needle in the Haystack • Data mining refers to the new genre of BI tools used to sift through the mass of raw data. • DM applications should be able to process – TEMPORAL (Time studied) and – SPATIAL (Organism, organ, cell type etc) data. – The gained ‘knowledge’ to reprocess data. – Data using techniques beyond Bayesian (similarity search) methods. • An extension of DM is the concept of ‘KNOWLEDGE DISCOVERY’, which open up new avenues of research with new questions and different perspectives. Commercial Structural Genomics Initiatives • IBM (Blue Gene project: 2000) – Computational protein folding • Geneformatics (1999) – Modeling for identifying active sites • Prospect Genomics (1999) – Homology modeling • Protein Pathways (1999) – Phylogenetic profiling, domain analysis, expression profiling • Structural Bioinformatics Inc (1996) – Homology modeling, docking