Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Exome sequencing wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Non-coding DNA wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
SNP genotyping wikipedia , lookup
Molecular evolution wikipedia , lookup
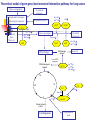
Genome-Wide Association Studies (GWAS) Slides 1-35 modified from: http://webcache.googleusercontent.com/search?q=cache:7CMIHVNXPGMJ:www.ph.ucla.edu/epi/fac ulty/zhang/Webpages/zhang/courses/epi243_07/lectures/GenomeWide_Association_Studies_(GWAS).ppt+&cd=2&hl=en&ct=clnk&gl=us&client=safari Association Studies of Genetic Factors 1st generation Very small studies (<100 cases) Usually not epidemiologic study design; 1-2 SNPs 2nd generation Small studies (100-500 cases) More epi focus; a few SNPs 3rd generation Large molecular epi studies (>500 cases) Proper epi design; pathways 4th generation Consortium-based pooled analyses (>2000 cases) GxE analyses 5th generation Post-GWS studies Boffeta, 2007 International Lung Cancer Consortium (ILCCO) Wichmann McLaughlin Schwarts Wild Boffetta Harris Goodman Risch Kiyohara Brennan Benhamou Wiencke Christiani Zhang Stucker Yang Tajima Landi Berwick Hong Vineis Lan Chen Lazarus Spitz Thun Le Marchand 3 cohort studies 17 population based case-control studies 13 hospital based case-control studies 2 studies with mixed controls 1 cross-sectional study Issues in genetic association studies Many genes Many SNPs ~25,000 genes, many can be candidates ~12,000,000 SNPs, ability to predict functional SNPs is limited Methods to select SNPs: Only functional SNPs in a candidate gene Systematic screen of SNPs in a candidate gene Systematic screen of SNPs in an entire pathway Genomewide screen Systematic screen for all coding changes Introduction A genome-wide association study is an approach that involves rapidly scanning markers across the complete sets of DNA, or genomes, of many people to find genetic variations associated with a particular disease. Once new genetic associations are identified, researchers can use the information to develop better strategies to detect, treat and prevent the disease. Such studies are particularly useful in finding genetic variations that contribute to common, complex diseases, such as asthma, cancer, diabetes, heart disease and mental illnesses. http://www.genome.gov/20019523 Definition of GWAS A genome-wide association study is defined as any study of genetic variation across the entire human genome that is designed to identify genetic associations with observable traits (such as blood pressure or weight), or the presence or absence of a disease (such as cancer) or condition. Potential of GWAS Whole genome information, when combined with epidemiological, clinical and other phenotype data, offers the potential for increased understanding of basic biological processes affecting human health, improvement in the prediction of disease and patient care, and ultimately the realization of the promise of personalized medicine. In addition, rapid advances in understanding the patterns of human genetic variation and maturing high-throughput, cost-effective methods for genotyping are providing powerful research tools for identifying genetic variants that contribute to health and disease. Potential of GWAS Selection of SNPs (Genome-wide association studies) Molecular Analytical Highest requirements: Data management, automation Advantages Higher requirements: Affymetrix and Illumina No biological assumptions and can identify novel genes/pathways Excellent chance to identify risk alleles Utility in individual risk assessment Disadvantages High costs Concern of multiple tests SNP Selection Affymetrix® Genome-Wide Human SNP Array The new Affymetrix® Genome-Wide Human SNP Array 6.0 features 1.8 million genetic markers, including more than 906,600 single nucleotide polymorphisms (SNPs) and more than 946,000 probes for the detection of copy number variation. The SNP Array 6.0 represents more genetic variation on a single array than any other product, providing maximum panel power and the highest physical coverage of the genome. The need for GWA Current understanding of disease etiology is limited Current understanding of functional variants is limited Xu JF, 2007 Therefore, the focusing on nonsynonymous changes is not sufficient Results from linkage studies are often inconsistent and broad Therefore, candidate genes or pathways are insufficient Therefore, the utility of identified linkage regions is limited GWA studies offer an effective and objective approach Better chance to identify disease associated variants Improve understanding of disease etiology Improve ability to test gene-gene interaction and predict disease risk GWA is promising Many diseases and traits are influenced by genetic factors Over 12 millions SNPs are known in the genome i.e., it is affordable to genotype a large number of SNPs in the genome Large numbers of cases and controls are available i.e., some SNPs will be directly or indirectly associated with causal variants The cost of SNP Genotyping is reduced i.e., they are caused by sequence variants in the genome i.e., there is statistical power to detect variants with modest effect When the above conditions are met… …associated SNPs will have different frequencies between cases GWA is challenging Many diseases and traits are influenced by genetic factors But probably due to multiple modest risk variants They confer a stronger risk when they interact True associated SNPs are not necessary highly significant Too many SNPs are evaluated Single studies tend to be underpowered Xu, 2007 False positives due to multiple tests False negatives Considerable heterogeneity among studies Phenotypic and genetic heterogeneity False positives due to population stratification Genome coverage Two major platforms for GWA Illumina: HumanHap300, HumanHap550, and HumanHap1M Affymetrix: GeneChip 100K, 500K, 1M, and 2.3M Genome-wide coverage The percentage of known SNPs in the genome that are in LD with the genotyped SNPs Calculated based on HapMap (Haplotype map) Xu, 2007 http://hapmap.ncbi.nlm.nih.gov/downloads/nature02168.pdf Calculated based on ENCODE Encyclopedia of DNA Elements identify all functional elements in the human genome. https://www.ncbi.nlm.nih.gov/pubmed/21037257?dopt=Abstract Strategies for pre-association analysis Quality control Filter SNPs by genotype call rates Filter SNPs by minor allele frequencies Filter SNPs by testing for Hardy-Weinberg Equilibrium (p + q)2 = p2 + 2pq + q2 = 1 Data Analysis Single SNP analysis using prespecified genetic models 2 x 3 table (2-df) Additive model (1-df), and test for additivity All possible genetic models (recessive, dominant) Data Analysis Haplotype analysis Gene-gene and gene-environment interactions Interaction with main effect Logistic regression Interaction without main effect: data mining Classification and recursive tree (CART) Multifactor Dimensionality Reduction (MDR) Sample size needs as a function of genotype prevalence and OR for main effects Boffeta, 2007 False Positives False positives: too many dependent tests Adjust for number of tests Bonferroni correction Nominal significance level = study-wide significance / number of tests Nominal significance level = 0.05/500,000 = 10-7 Effective number of tests Take LD into account Permutation procedure Permute case-control status Mimic the actual analyses Obtain empirical distribution of maximum test statistic under null hypothesis False Positives False discovery rate (FDR) Expected proportion of false discoveries among all discoveries Offers more power than Bonferroni Holds under weak dependence of the tests False Positives Bayesian approach Taking a priori into account, False-Positive Report Probability (FPRP) Confirmation in independent study populations The approach may limit the number of false positives Confirmation is needed to dissect true from false positives Replication, examine the results from the 2nd stage only Joint analysis, combining data from 1st stage with 2nd stage Multiple stages Issues of GWAS Population stratification Multiple Testing: False Positives Gene-Environmental Interaction High Costs Kingsmore, 2008 Kingsmore, 2008 Hypothesis The overall hypothesis is that multiple sequence variants in the genome are associated with the risk of lung cancer among non-smokers. Specifically, we hypothesize that a number of common nonsmoking lung cancer risk-modifying SNPs are in strong LD with the SNPs arrayed on the 500K GeneChip®. Theoretical model of gene-gene/environmental interaction pathway for lung cancer Tobacco consumption Occupational Exposures Environmental Carcinogens / Procarcinogens Exposures Ile105Val Ala114Val Environmental Exposure Null GSTP1 GSTM1 CYP1A1 MspI Ile462Val Tyr113His His139Arg PAHs, Xenobiotics, Arene, Alkine, etc Detoxified carcinogens Active carcinogens Pro187Ser mEH mEH NQO1 DNA damage repaired DNA Damage Tyr113His His139Arg Normal cell Defected DNA repair gene If DNA damage not repaired XRCC1 Arg194Trp, Arg399Gln, Arg280His M G1 G2 P53 P16 S G0 G870A Arg72Pro Ala146Thr Cyclin D1 If loose cell cycle control Carcinogenesis Programmed cell death Figure 1. The effects of SNPs on the Risk of Lung Cancer among Smokers and Non-smokers 8 OR 7 6 5 Smokers Non-Smokers ETS Exp Non ETS Exp 4 3 2 1 0 BRCA1 CHEK1 XRCC3 INFG IL-10 ALDH2 Flow cytometry analysis Facsalibur sorting Fortessa cytometer Excitation Optics The excitation optics consist of multiple fixed wavelength lasers, beam shaping optics, and individual pinholes which result in spatially separated beam spots. A final lens focuses the laser light into the gel-coupled cuvette flow cell. Since the optical pathway and the sample core stream are fixed, alignment is constant from day to day and from experiment to experiment. Collection Optics Emitted light from the gel-coupled cuvette is delivered by fiber optics to the detector arrays. The collection optics are set up in patented octagon- and trigon-shaped optical pathways that maximize signal detection resulting from each laser illuminated beam spot. Bandpass filters in front of each PMT allow spectral selection of the collected wavelengths. Importantly, this arrangement allows filter and mirror changes within the optical array to be made easily and requires no additional alignment for maximum signal strength. The analyzer can be configured with up to 5 lasers to detect up to 20 parameters simultaneously to support ever increasing demands in multicolor flow cytometry. A wide range of up to 34 laser choices is available as excitation sources, including blue, red, violet, yellow-green, and UV FACSAria Three lasers provide excitation at 407, 488, and 633 nm for analysis of up to 10 fluorescence channels plus forward and side scatter Digital electronics Sort up to four populations simultaneously Spectral overlap http://bitesizebio.com/13696/introduction-to-spectral-overlap-and-compensation-flow-cytometry-protocol/ Compensation is the process of correcting the spillover from our primary signal in each secondary channel it is measured in. Figure 2: Fluorescein emission profile with two filters overlaid. The standard filter for fluorescein is a 530/30 filter. This filter allows light between 515-545 nm to pass through the filter. The second filter, 585/42, is a common filter for the fluorescent molecule phycoerythrin (PE) and allows light between 564-606 nm to pass. The overlap of the fluorescein molecule into the PE detector indicates that approximately 12% of the fluorescein molecule is being measured in the PE detector. Figure generated using the Invitrogen spectral viewer. https://www.thermofisher.com/us/en/home/life-science/cell-analysis/labeling-chemistry/fluorescence-spectraviewer.html