Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Amino acid synthesis wikipedia , lookup
Catalytic triad wikipedia , lookup
Genetic code wikipedia , lookup
Epitranscriptome wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Non-coding DNA wikipedia , lookup
Protein structure prediction wikipedia , lookup
Metalloprotein wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Biosynthesis wikipedia , lookup
Biochemistry wikipedia , lookup
Microbial metabolism wikipedia , lookup
Homology modeling wikipedia , lookup
Enzyme inhibitor wikipedia , lookup
Structural alignment wikipedia , lookup
Point mutation wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
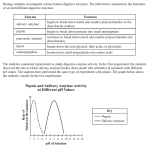
Diversity in the Structure and Function of Amylase Kim Gernert and Vedham Karpakakunjaram Target audience: Students enrolled in Principles of Biology I (BI 107) and II (BI 108) Background Alpha amylase: http://www.rcsb.org/pdb/101/motm.do?momID=74 Alpha-amylase begins the process of starch digestion. It takes starch chains and breaks them into smaller pieces with two or three glucose units. Human salivary amylase is used in one of our lab modules, so students are familiar with the enzyme and its function. Questions • Given organisms from three domains with diverse lifestyles and study sequence differences and their effect on enzyme’s structure and function. • Are the structures of amylase different across organisms? • Relate the identity and percentage similarities in sequences based on clustering in the phylogenetic tree. • Relate the conserved regions of the alignment to the secondary structure of amylase. • Identify the active site and locate the critical residues and find their conservation of the sequences in the alignment. • Study the binding of other molecules in the active site including inhibitors. • How do mutations in amylase modify their structures? • How do these modifications alter the enzyme’s function? Homo_sapie ALGKDYVRSKIAEYMNHLID-IGVAGFRIDASKHMWPGDIK--------------AILDK n3VM5_Meda ALEKDYVRGKVADFMNKLID-MGVAGFRVDACKHMWPGDLD--------------NVYRR Tenebrio_m NQGSDYVRGVLIDYMNHMID-LGVAGFRVDAAKHMSPGDLS--------------VIFSG sp|P25718| GTFHGGDLRGLTNKLDYLQQ-LGVNALWISAPFEQIHGWVGGGTKGDFPHYAYHGYYTQD Saccharomy RTEDSDVASVFNSWVKDFVGNYSIDGLRIDSAKHVDQGFFP-------------DFVSAS Oryza_sati DHLNKRVQRELIGWLDWLKMDIGFDAWRLDFAKGYS----------------------AD Pyrococcus THELVYERGWLKEFFDRISSDDKINLMLYSEYLSKFRPKGLVYLPIASYFEMSEWSLPAR . .. : . . Homo_sapie LHNLNSNW-FPEGSKPFIYQEVID----LGGEPIKSSDYFGNGRVTEFKYGAK—LGTVI n3VM5_Meda LNNLNTKW-FPGGSRPFIFQEVID----LGGEPITTGEYVGLGRVTEFKYGAR—LGELF Tenebrio_m LKNLNTDYGFADGARPFIYQEVID----LGGEAISKNEYTGFGCVLEFQFGVS—LGNAF sp|P25718| WTNLDANMGNEADLRTLVDSAHQRGIRILFDVVMNHTGYATLADMQEYQFGALYLSGDEV Saccharomy GVYSVGEVFQGDPAYTCPYQNYIP----------GVSNYPLYYPTTRFFKTTDSSSSELT Oryza_sati MAKIYIDATEPSFAVAEIWTSMAN----------GGDGKPNYDQNAHRQELVNWVDRVGG Pyrococcus QAKLFFEFIKKLKELNLFEKYRIFVRG------GIWKNFLYKYPEGNYMHKRMLMLSKLL Tools: • Amino acid sequences (PDB, EBI) from many diverse organisms to be provided for students to select about 5-6 organisms representing the three domains. Hyperthermophilic Archeae, parasitic and mutualistic bacteria, unicellular and multicellular eukaryotes. • Compare the sequences with alignments from CLUSTALW and construct phylograms in: www.phylogeny.fr • Use NCBI (http://www.ncbi.nlm.nih.gov/) for comparing the identity and percent similarities in the sequences across organisms in order to synthesize the information from phylogenetic trees. • Use Chimera (www.cgl.ucsf.edu/chimera) to visualize: conserved and modified regions, especially around the active site. the binding of substrate, inhibitors to determine the nature of inhibition (competitive) Provide sequences of mutations to be compared against normal sequences for identification of conserved and modified regions, and to predict modifications in the mutant enzyme’s function, if any.