Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Multi-state modeling of biomolecules wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Magnesium transporter wikipedia , lookup
Expression vector wikipedia , lookup
Genetic code wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Metalloprotein wikipedia , lookup
Interactome wikipedia , lookup
Biochemistry wikipedia , lookup
Western blot wikipedia , lookup
Protein purification wikipedia , lookup
Homology modeling wikipedia , lookup
Point mutation wikipedia , lookup
Two-hybrid screening wikipedia , lookup
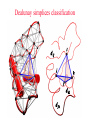
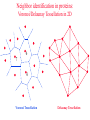
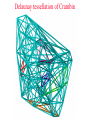
CASB workshop, 9/23/10 Protein Function Analysis using Computational Mutagenesis Iosif Vaisman Laboratory for Structural Bioinformatics proteins.gmu.edu Department of Bioinformatics and Computational Biology Dealunay simplices classification Protein representation (Crambin) Neighbor identification in proteins: Voronoi/Delaunay Tessellation in 2D Delaunay simplex is defined by points, whose Voronoi polyhedra have common vertex Delaunay simplex is always a triangle in a 2D space and a tetrahedron in a 3D space Voronoi Tessellation Delaunay Tessellation Neighbor identification in proteins: Voronoi/Delaunay Tessellation in 2D 6 7 6 Voronoi Tessellation Delaunay Tessellation Delaunay tessellation of Crambin Delaunay Tessellation of Protein Structure D (Asp) Cα or center of mass Abstract each amino acid to a point Atomic coordinates – Protein Data Bank (PDB) A22 L6 D3 F7 G62 K4 S64 R5 C63 Delaunay tessellation: 3D “tiling” of space into non-overlapping, irregular tetrahedral simplices. Each simplex objectively defines a quadruplet of nearest-neighbor amino acids at its vertices. Compositional propensities of Delaunay simplices k q ijkl l i j fijkl log pijkl f- observed quadruplet frequency, pijkl = Caiajakal, a - residue frequency AAAA: C = 4! / 4! = 1 AAAV: C = 4! / (3! x 1!) = 4 AAVV: C = 4! / (2! x 2!) = 6 AAVR: C = 4! / (2! x 1! x 1!) = 12 AVRS: C = 4! / (1! x 1! x 1! x 1!) ) = 24 C 4! n (t !) i i Counting Quadruplets • assuming order independence among residues comprising Delaunay simplices, the maximum number of all possible combinations of quadruplets forming such simplices is 8855 C D E F 20 4 4845 C C D E 19 20 2 3420 C C D D 20 2 190 C C C D 20 19 380 C C C C 20 20 8855 Log-likelihood of amino acid quadruplets with different compositions Log-likelyhood ratio 1.5 1.0 0.5 0.0 -0.5 -1.0 -1.5 1 2 3 4 5 6 7 8 9 ... CCCC CCCY CCHH CCCG CCCH CCCW CCCS CCCQ CCCF ... 2000 3.081003 2.13004 1.960814 1.782267 1.742759 1.724275 1.724275 1.657329 1.621613 ... 8000 6000 4000 ... 8343 8344 8345 8346 8347 8348 8349 8350 8351 ... CDDL IRRV AEYY KKRV CKRS CEKP HKKS CGLR ACKN ... -0.90166 -0.90217 -0.90535 -0.95081 -0.96133 -0.98433 -0.98472 -1.14737 -1.16297 Delaunay simplices with distinct composition Log-likelihood of amino acid quadruplets Log-likelihood of amino acid quadruplets Computational Mutagenesis Methodology • Observations: • Relatively few mutant and wt structures of same protein have been solved • Tessellations of mutant and wt protein structures are very similar or identical • Approach: • Obtain topological score (TSmut) and 3D-1D potential profile vector (Qmut) for any mutant protein by using the wt structure tessellation as a template • Simply change the residue label at a given point(s) and re-compute s(R,D,A,L) A22 s(I,D,A,L) L6 s(R,G,F,L) Mutation F7 D3 (R5 I5) s(R,D,K,S) G62 K4 S64 R5 A22 L6 s(I,G,F,L) D3 F7 s(I,D,K,S) G62 K4 S64 s(R,S,C,G) C63 (TSwt, Qwt) I5 s(I,S,C,G) C63 (TSmut, Qmut) Computational Mutagenesis Methodology • Scalar “Residual Score” of a mutant: (mutant – wt) topological score difference = TSmut – TSwt (empirical measure of relative structural change due to mutation) • Vector “Residual Profile” of a mutant: R = Qmut – Qwt = (mutant – wt) 3D-1D potential profile difference (environmental perturbation score at every position in structure) • Denote R = < EC1, EC2, EC3,…, ECN > ECi = qi,mut – qi,wt = relative Environmental Change at position i • Geometric property: If mutant is due to a single substitution at position j, then ECj ≡ mutant residual score (“epicenter” of impact) • The only other nonzero EC components correspond to neighboring positions that participate in simplices with j Approach 1: Protein Topological Score (TS) • Obtained by summing the log-likelihood scores of all simplicial quadruplets defined by the protein tessellation • Global measure of protein sequence-structure compatibility • Total (empirical or statistical) potential of the protein TS = ∑î s(î), sum taken over all simplex quadruplets î in the entire tessellation. s(R,D,A,L) A22 L6 s(R,G,F,L) D3 F7 s(R,D,K,S) G62 K4 S64 R5 s(R,S,C,G) C63 Close-up view of only the four simplices that use R at position 5 as a vertex (hypothetical) Approach 2: Residue Environment Scores • For each amino acid position, locally sum the log-likelihood scores s(i,j,k,l) of only simplex quadruplets that include it as a vertex s(R,D,A,L) A22 L6 s(R,G,F,L) D3 F7 s(R,D,K,S) G62 K4 S64 R5 Example: q5 = q(R5) = ∑(i,j,k,l) s(i,j,k,l), sum over all simplex quadruplets (i,j,k,l) that include amino acid R5 s(R,S,C,G) C63 • The scores of all amino acid positions in the protein structure form a 3D-1D Potential Profile vector Q = < q1, q2, q3,…,qN > (N = length of primary sequence in solved structure) Reversibility Analysis S1,E1 ‘reference’ PDB S1,E2 Calculated Mutant Forward Mutation S2,E2 Mutant PDB S2,E1 Calculated ‘reference’ Reverse Mutation Reversibility of mutations (T4 lysozyme) Protein Mutation 1l63 180l T26E E26T 1l63 123l 1l63 1cu3 A82S S82A V87M M87V Score change -2.49 2.01 4 3 1.49 -1.49 2 1 -0.28 0.22 0 -4 1l63 138l A93C C93A -1.98 1.78 -3 -2 -1 0 1 2 -1 -2 R2 = 0.9886 1l63 1goj T152S S152T -1.08 1.12 -3 3 Reversibility Analysis Reversibility Analysis Reverse mutation potential score difference 15 y = -0.9918x - 0.0251 R2 = 0.9742 10 5 0 (4pnp,1ao9) -5 -10 -15 -15 -10 -5 0 5 Forw ard m utation potential score difference 10 15 Functional Effects of Amino Acid Substitutions • Change in protein stability: • Effect on melting temperature: ΔTm = Tm (mutant) – Tm (wt) • Effect on thermal denaturation: ΔΔG = ΔG (mutant) – ΔG (wt) • Effect on denaturant denaturation: ΔΔGH2O = ΔGH2O (mutant) – ΔGH2O (wt) • Change in protein activity: • Mutant enzymatic activity relative to wt • Mutant strength of DNA binding relative to wt • Disease potential of human coding nsSNPs • Neutral polymorphism or disease-associated mutation? • For protein targets of inhibitor drugs: • Continued susceptibility or (degree of ) resistance that patients with the mutant protein have to the inhibitor • Inhibitor binding energy to mutant target relative to wt Examples ofExperimental Mutagenesis Data • HIV-1 protease (99 aa’s): 536 single point mutants (at least one at each position) – • Bacteriophage T4 lysozyme (164 aa’s): 2015 single point mutants (12-13 at each position except first) – • D. D. Loeb et al., Nature 340, 397 (1989). D. Rennell et al., J. Mol. Biol. 222, 67 (1991). E. coli lac repressor (360 aa’s): 4041 single point mutants (12-13 at positions 2 to 329) – P. Markiewicz et al., J. Mol. Biol. 240, 421 (1994). Example: HIV-1 Protease (PR) HIV-1 PR Dataset Example: Residual Profiles of 536 Experimental Mutants … … Experimental Mutants: Residual Scores Elucidate the Structure-Function Relationship 536 HIV-1 protease mutants 630 hIL-3 mutants 4041 lac repressor mutants 371 gene V protein mutants Universal Model Approach: 8635 Experimental Mutants from 7 Proteins Universal Model Approach: 980 Experimental Mutants from 20 Proteins Mean Residual Score _ 0.6 0.3 0 -0.3 -0.6 -0.9 Increased Decreased Mutant Protein Stability Change Structure-Function Correlation Based on Residual Scores: nsSNPs • 1790 nsSNPs corresponding to single amino acid substitutions in several hundred proteins with tessellatable structures • Function: 1332 nsSNPs associated with disease; 458 neutral • Data obtained from Swiss-Prot and HPI Structure-Function Correlation Based on Residual Scores: Drug Susceptibility Mean Residual Score_ 0.00 -0.20 -0.40 NFV: -0.26 SQV: -0.19 IDV: -0.48 RTV: 0.09 APV: -0.49 LPV: -0.41 ATV: 0.05 Average: -0.28 -0.60 -0.80 NFV: -0.18 SQV: -1.05 IDV: -0.93 RTV: -0.87 APV: -0.80 LPV: -0.78 ATV: -0.72 Average: -0.77 -1.00 NFV: -1.10 SQV: -1.23 IDV: -1.00 RTV: -0.99 APV: -1.24 LPV: -1.04 ATV: -1.17 Average: -1.09 -1.20 Sensitive Intermediate Resistant Susceptibility to HIV-1 Protease Inhibitors Algorithm Performance: 2015 T4 Lysozyme Mutants Learning Curves for HIV-1 protease and T4 lysozyme mutants Real-World Application: T4 Lysozyme Predictions • Experimental data (not part of training set) obtained from ProTherm database • Result: predictions match experiments for 30/35 (~86%) of the mutants T4 Lysozyme Mutational Array Training set mutants (n = 2015) Active Inactive Predicted test set mutants (n = 1101) Active Inactive GVP Mutational Array Support Vector Regression Capriotti et al. SVM regression (for comparison): r = 0.71, Standard Error = 1.3 kcal/mol, y = 0.5223x – 0.4705 Conclusions • Computational mutagenesis derived from a four-body, knowledge-based statistical potential uniquely characterizes each protein mutant using both sequential and structural features • Attributes correlate well with mutant function valuable for developing accurate machine learning based predictive models Acknowledgements Structural Bioinformatics Laboratory (GMU): Collaborators: Tariq Alsheddi David Bostick Andrew Carr Sunita Kumari Yong Luo Majid Masso John Grefenstette (GMU) Curt Jamison (GMU) Dmitri Klimov (GMU) Dan Carr (GMU) Estela Blaisten (GMU) Vladimir Karginov (IB) Ewy Mathe Olivia Peters Vadim Ravich Greg Reck Todd Taylor Bill Zhang (structure alignment) (topological similarity) (functional sites, visualization) (structural genomics) (evolutionary structure analysis) (mutagenesis, HIV-1 protease, LAC repressor, T4 lysozyme, SNP) (mutagenesis, p53) (protein-protein interfaces) (HIV RT mutagenesis) (hydration potentials, amyloids) (statistical potentials, secondary structure, topology, protein stability) (mutagenesis, BRCA1) Unpublished data: Clyde Hutchison (UNC) Ron Swanstrom (UNC) Funding: NSF NIH-Innovative Biologics GMU-INOVA Research Fund Evaluating Algorithm Performance • Overall goal: Develop model with known examples to accurately predict class (or value) of instances that have not yet been assayed experimentally (potentially great savings of time and money) • Ideal situation: split large original dataset into 3 subsets o o o o Training set (learn model) Validation set (optimize model by tweaking model parameters) Test set (evaluate model on new data not used to develop model) Errors measured at each step (resubstitution, validation, generalization) • Approaches: Tenfold cross-validation (10-fold CV); leave-one-out CV (i.e., jackknife or N-fold CV, N = dataset size); % split (e.g., use only 2/3 for training, 1/3 held out for testing) Evaluating Algorithm Performance • 10-fold CV o Randomly split the dataset instances into 10 equally-sized subsets o Hold-out subset 1; combine subsets 2-10 into one training set for learning a model; use trained model to predict classes of instances in subset 1 o Repeat previous step 9 more times (e.g., hold-out subset 2, combine subsets 1 and 3-10 together to train a model, use model to predict subset 2, etc) o We end up with 10 models, each trained using 90% of the original dataset, and each used to predict the held-out 10% subset. o In the end, each instance has one class prediction – compare to actual class • LOOCV (leave-one-out CV, jackknife, or N-fold CV) o Similar to above, but each subset contains only 1 instance o Deterministic – no randomness to which instances are grouped as subsets o Overall prediction accuracy provides rough idea of how a model trained with the full dataset will perform • % split (self-explanatory) Evaluating Algorithm Performance • Assume instances belong to two generic classes (Pos/Neg) • Results of comparing predictions with actual classes based on the approaches described (10-fold CV, LOOCV, % split) can be summarized in a confusion matrix: Predicted as Actual class Pos Neg Pos TP FN Neg FP TN • Classification performance measures: accuracy = (TP+TN) / (TP+FP+TN+FN); sensitivity = TP / (TP+FN); specificity = TN / (TN+FP); precision = TP / (TP+FP); BER = 0.5 × [FP / (FP+TN) + FN / (FN+TP)]; MCC = (TP×TN – FP×FN) / (TP+FN)(TP+FP)(TN+FN)(TN+FP); AUC = area under ROC curve (plot of sensitivity vs. 1 – specificity) • For regression models: correlation coefficient, standard error ROC Curve • Plot of true positive rate (sensitivity) versus false positive rate (1 – specificity) in the unit square • AUC = probability that classifier will rank a randomly chosen positive instance higher than a randomly chosen negative one • AUC ~ 0.5 (ROC close to diagonal line joining points (0,0) and (1,1)) suggests no signal in dataset and that trained model is not likely to perform any better than random guessing • AUC = 1 (piecewise linear ROC joining (0,0) to (0,1) and (0,1) to (1,1)) indicates a perfect classifier