Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Adaptive immune system wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Neonatal infection wikipedia , lookup
Molecular mimicry wikipedia , lookup
Infection control wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
Innate immune system wikipedia , lookup
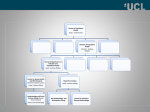
Mutation and Control of the Human Immunodeficiency Virus Robert F. Stengel Princeton University and Institute for Advanced Study Princeton, NJ e-mail: [email protected] Abstract - We examine the dynamics of infection by the human immunodeficiency virus (HIV), as well as therapies that minimize viral load, restore adaptive immunity, and use minimal dosage of anti-HIV drugs. Virtual therapies for wild-type infections are demonstrated; however, the HIV infection is never cured, requiring continued treatment to keep the condition in remission. With high viral turnover and mutation rates, drug-resistant strains of HIV evolve quickly. The ability of optimal therapy to contain drugresistant strains is shown to depend upon the relative fitness of mutant strains. Index Terms – Human immunodeficiency virus, immune system dynamics, optimal control. I. INTRODUCTION Tens of millions of people have been infected by the human immunodeficiency virus (HIV) since it was first recognized in the early 1980s, and more than 20 million have died from ensuing disease [1-4]. The virus attacks CD4-presenting cells -- helper T cells, macrophages, dendritic cells, eosinophils, microglia, and natural killer cells -- mainstays in the body’s immune response to pathogenic attack. Without treatment, immune cells and lymphatic tissue are destroyed, and the infection evolves into acquired immune deficiency syndrome (AIDS), an inevitably fatal condition for all but a few [5]. While AIDS itself is rarely the cause of death, the body becomes vulnerable to a host of potentially lethal opportunistic infections and malignancies, including pneumonia, tuberculosis, dementia, bacterial sepsis, lymphoma, and Kaposi’s sarcoma. With multi-drug, highly active antiretroviral therapy (HAART), the rate of HIV infection can be dramatically slowed; fatalities brought about by the disease have plummeted since the inception of HAART [6]. presented at the 13th Yale Workshop on Adaptive and Learning Systems, Yale University, New Haven, CT, May 30 - June 1, 2005. Copyright 2005 by Robert F. Stengel. All rights reserved. Nevertheless, HAART is not a cure because current drug regimens cannot eliminate diverse HIV strains in a broad population [6-13]. Furthermore, HAART can lead to serious and discomforting side effects. Cardiovascular disease, hepatitis, pancreas damage, glucose intolerance, kidney stones, rashes, depression, fat accumulation and redistribution, nausea, diarrhea, pain or numbness in the feet, mouth inflammation, blurred vision, headache, dizziness, anemia, weakness, insomnia, and adverse interaction with non-HIV drugs are not uncommon [14]. There are a number of reasons why the human immunodeficiency virus cannot be defeated with current therapy. The virus targets different immune-system cell types that are capable of replicating the virus; treatments that inhibit HIV replication in one cell type may not be as effective for another. Once infected by a single HIV particle (or virion), an immune cell may produce over a thousand copies of the virion before the cell is destroyed; hence, unchecked infection is exponentially unstable. The virus is short-lived but prolific, with maximum daily turnover rates measured in the billions of virions. Because all encounters between cells and molecules within the body’s circulatory systems are stochastic, both infection and therapeutic effect are never certain. The virus sequesters itself in a variety of reservoirs throughout the body, some of which are physically resistant to therapy. Long-term remission requires continued stable control over the immune cells that are vulnerable to infection for the remainder of the patient’s life. There is no assurance that the last virion will not replicate unless the therapy induces stable decay to the HIV production process [15]. Perhaps most important, the infection is able to adapt to maintain its virulence in the presence of natural immune response and drug therapy. HIV is a retrovirus that transmits the genetic code contained in its dual strands of RNA by reverse transcription to cDNA [1]. This process is not monitored and controlled by the exonuclease proof reading that occurs in forward transcription from DNA to RNA. Thus, the DNA that the virus splices into the host cell’s genome is highly variable. In the cell’s subsequent “normal” transcription, many mutants of the virus are replicated, and some of these are likely to resist therapy. Mutation and Control of the Human Immunodeficiency Virus Together with a high production rate, the continuing appearance of diverse mutants is assured. In the majority of patients, the mutant virus destroys the immune system and eludes treatment, progressing to AIDS and, finally, death. In the remainder of the paper, we summarize HIV infection and immune response, present a mathematical model of HIV infection with drug-resistant mutation, and demonstrate the effects of mathematically optimal therapy. The virtual therapy that we derive is optimal in the sense that it minimizes the viral load and infected T cell count, trading off the beneficial effects of reduced concentrations of the virus and infected cells against the adverse side effects of therapy. We see that normal competition between wild-type and less-fit variant HIV strains plays an important role in providing such control; above a certain fitness level, the drug-resistant strain cannot be controlled. Opportunities to apply adaptive control concepts are limited because high-fitness mutation defeats the efficacy of known therapeutic agents. II. INFECTION AND IMMUNE RESPONSE The HIV infection of CD4-presenting cells begins with transfer of the virus across the mucous membranes of sexual partners, transfusion of HIV-infected blood, transplantation of HIV-infected tissue, pre- or post-natal infection of a child by its mother, or injection using an infected needle. Macrophages and helper T cells (or Th cells) are generally among the first cells to be infected. The infection is then passed to other immune-system cells primarily within lymphoid tissue [16]. Th cells are depleted, and the functions of lymphoid organs are disrupted over the course of HIV infection [1, 5, 17, 18]. Once the HIV particle is fused to the target CD4 cell, the cell membrane is breached, and the contents of the virion enter the target [26]. Each viral RNA strand is accompanied by reverse transcriptase, an enzyme that facilitates the production of a corresponding cDNA strand, and integrase, an enzyme that aids the integration of the cDNA into the target cell’s genome. This integrated strand of DNA is called a provirus, and the infected cell is a proviral cell. Transcription of the modified DNA genome to its RNA image cannot occur unless the CD4 cell has recognized a foreign antigen from the HIV, another pathogen, or immunizing vaccine directed at another pathogen. Upon activation, transcription of RNA derived from the original and proviral DNA is initiated [19], and the cell becomes productively infected, that is, it can produce new copies of HIV particles. Transcription is followed by translation of the new RNA to original and HIV proteins. Through the action of a protease enzyme also carried in the infecting virion, proteins essential to creating new virions are produced. The HIV RNA and proteins move to the cell membrane, assemble into a viable unit, and “bud” from the target cell 2 as a new virion. This process occurs many times in a productively infected cell, producing exponential growth in the number of HIV particles. Ultimately, the cell undergoes programmed cell death (apoptosis), disintegrating and releasing additional HIV product into the compartment. Although there is a multiplicity of T-cell types involved in HIV infection, activated Th cells, which have recognized a foreign antigen, are the primary producers of and targets for HIV particles [20]. Most Th cells are infected and reside in lymphoid tissue [21], where they can be observed only by biopsy. Just 1-2% of infected cells reside in the periphery, where observation by blood test is possible; hence, the balance between cell destruction and impaired production that leads to depletion over time is not well understood. While Th cells produce most of the new virus, eosinophils [22], natural killer cells [64], microglia [23, 24], and macrophages that are within the central nervous system or have been co-infected by opportunistic pathogens [23, 25] can be productively infected, and lymphoid tissue can harbor HIV. Other immune system cell types may be infected but not killed by HIV, producing long-term reservoirs for the virus [22, 26, 27]; however, they generally do not present antigens to the B and T cells of the adaptive immune system [19] and, therefore, play a secondary role in the dynamics of HIV infection. The immune system recognizes and mounts a strong response to HIV infection through innate, humoral, and adaptive pathways [16, 28]. However, the response is not uniformly protective, and chronic or hyper-activation of the immune system is associated with poor clinical outcome [29, 30]. During the first weeks following infection, both CD4 and CD8 T cells proliferate at a high rate, there is a sharp drop in viral load, and antibodies for the virus’s antigens build to measurable levels in the blood. The innate response is not specific to the intruder’s antigen; it destroys virions and infected cells that are recognized as “non-self.” The humoral response is guided by antigen-specific antibodies, which bind the antigen and present a constant peptide region to invariant receptors on phagocytic cells. The adaptive response is spearheaded by activated killer (CD8) T cells, which require the help of Th cells (the principal target of infection) to recognize foreign antigen [19]. Killer T cells initially destroy large numbers of HIV-infected cells [15, 19, 29, 31-33], but the depletion of helper T cells over the course of time degrades the ability of killer T cells to destroy infected cells. The cytotoxic effects of antigen-specific CD8 T cells eliminate the HIV strains of early infection, but this action renders the trained cells obsolete and clears the way for mutant strains expressing different antigens to prevail. HIV infection has a profound effect not only on the functions of T cells but on the lymphoid tissue in which they are created and mature, the bone marrow and thymus. The pro- Mutation and Control of the Human Immunodeficiency Virus duction of new T cells decreases, and the body is increasingly dependent on cell division in the periphery to maintain immune protection. Thus, “the HIV-1-specific response becomes part of the problem as well as part of the solution” [34]. The human immunodeficiency virus is uniquely capable of immune system judo, leveraging the system’s signaling and effecting mechanisms to work against itself. Signaling and effector cells are depleted, organs atrophy, and immune function is lost at all levels, leaving the patient susceptible to the opportunistic infections and malignancies that characterize AIDS. III. THERAPY, PERSISTENCE, AND MUTATION Because HIV has a high turnover rate, the main goal of current HIV therapy is to break the replication cycle. Fundamental damage to the immune system occurs during the first weeks of infection, when the diversity of virions is small [34]. A “hit early, hit hard” approach to therapy is more likely to minimize damage to lymphoid tissue and to eradicate a higher percentage of virulent particles than delayed treatment would [35-38]. With ten to a hundred million productively infected copies circulating at any given time, Th cell infection is not only the disease’s predominant feature, it is at the core of the immune system’s dynamic response to HIV. Current HIV drugs enter the cells’ nucleus and cytoplasm via passive diffusion, and they operate at chokepoints in the replicative sequence. Reverse transcriptase inhibitors (RTIs) are intended to prevent viral RNA from producing viral cDNA, thereby preventing a target cell’s genome from being modified. The HIV’s reverse transcriptase cannot distinguish between viral nucleosides and nucleoside-analogue RTIs (NRTIs), which contain a chain-terminating sequence that prevents the formation of complete HIV cDNA [39, 40]. Non-nucleoside RTIs (NNRTIs) use a different mechanism, blocking a binding pocket in the HIV reverse transcriptase to halt synthesis. Protease inhibitors (PIs) act farther downstream in the replication process, preventing protease from cleaving the long HIV peptide chain produced by translation into smaller proteins that are essential to fusing virions with new targets. A PI does not prevent the synthesis of viral components, but it does render the virions non-infectious. RTIs act primarily in newly infected cells, while PIs are also effective in chronically infected cells [41]. Dosage is a critical factor not only in treating the disease but in causing adverse side effects and creating selective pressure for mutation. PIs are generally less toxic than RTIs, and they are less likely to stimulate drug-resistant mutants. The principal failing of these drugs is that they are vulnerable to mutation in the HIV genome. The genome contains about 104 base pairs, and the point mutation rate is about one per 103-104 base-pair replications, or one to 3 ten per virion replication [35]. Given the broad nature of mutation [42], only a small percentage of mutant quasispecies1 are viable [44]. However, the number of new virions produced each day is large, and it is just a matter of time before strains resistant to any monotherapy (i.e., single-drug therapy) appear. Without therapy, wild-type strains dominate most mutant strains; however, with therapy, wild-type quasispecies are repressed, giving less-fit (but potentially lethal) mutants the opportunity to survive [45]. While a single point mutation in the reverse-transcriptase genomic sequence is sufficient to cause resistance to an NNRTI [46], multiple mutations are required to escape NRTIs or PIs [43, 47]. Furthermore, resistance to one drug in a class (e.g., NRTI, NNRTI, or PI) can produce cross-resistance to other drugs in the same class [47]. Consequently, monotherapy is susceptible to early drug resistance. Because HAART employs several drugs of different types, many mutations are required to defeat the therapy. Adding new classes of drugs to the HAART “cocktail,” such as attachment, fusion, maturation, and integrase inhibitors or adjuvant agents (which augment natural immune functions), should further increase the number of mutations required to defeat treatment [48-51]. Tailoring therapeutic profiles through a better understanding of HIV-immune system dynamics can improve overall efficacy and minimize side effects [52-54]. Preventative vaccines are desperately sought, but there is little hope that they will be available soon [55, 56]. IV. M ATHEMATICAL MODELS AND CONTROL OF HIV-IMMUNE SYSTEM DYNAMICS As noted in [57], mathematical models of HIV-immune system dynamics (or “host-pathogen interactions”) were proposed not later than 1986 [58], and the disease has become the subject of intense modeling efforts. A review of all of the models is beyond the present scope; however, we direct attention to a number of important papers. References [13, 32, 36, 59-88] investigate dynamic models of host-pathogen interactions; these papers compare and correlate response characteristics with biological and clinical observations. Several of the papers demonstrate computed effects of therapeutic alternatives, such as switching various drugs on or off during the course of infection. Systems and control theories are applied to the specification of therapeutic protocols in [8994], particularly through the use of open-loop optimization or stabilization via feedback. All of these models and design studies are based on ordinary differential equations, typically of order four or less. 1 A quasispecies is a family of strains that are genetically “close” if not identical [43]. Mutation and Control of the Human Immunodeficiency Virus 4 Wild-Type Model of Host-Pathogen Interaction The present paper takes the fourth-order model of [89] as a starting point, with modifications to the ways in which control effects are expressed, with significant changes to the cost function that is minimized by optimal control, and with the addition of a drug-resistant compartment. The four elements of the initial model's state represent concentrations of free, wild-type HIV particles (x1), uninfected Th cells (x2), proviral infected Th cells (x3), and productively infected Th cells (x4) in both the periphery and lymphoid organs. The model of host-pathogen dynamics (Fig. 1) is expressed by the equations, x˙1 = "a1 x1 " a2 x1 x 2 (1" u2 ) + a3 a4 x 4 (1" u1 ) # x + x3 + x4 & a x˙ 2 = 5 " a2 x1 x 2 (1" u2 ) " a6 x 2 + a7 %1" 2 ( x 2 (1+ u3 ) 1+ x1 a8 $ ' ˙x 3 = a2 x1 x 2 (1" u2 ) " a9 x 3 (1" u4 ) " a6 x 3 x˙ 4 = a9 x 3 (1" u4 ) " a4 x 4 terms in the equations. If an inhibitor takes a value of one (100% efficacy), it eliminates the term’s effect. When u3 takes a value of one, the concentration-limited rate of healthy Th cell production is doubled. Here, we illustrate monotherapies that employ either PIs or RTIs. A typical untreated wild-type response is shown in Fig. 2. The initial condition is taken at a point beyond sero-conversion where the immune system is still reducing viral load. Uninfected Th cell concentration is climbing (a good response), but so are the concentrations of infected cells, indicating that the HIV replication process is progressing. Over a simulated period of a year-and-ahalf, the concentrations of free virions and infected Th cells grow, while concentration of uninfected Th cells decays. Left unchecked, the patient generally progresses to AIDS when the total Th cell concentration falls below 200 cells per mm3 of blood. (1-4) ! where the form, rationale, and parameter values for the equations are given in [89]. The degree to which these equations reflect current knowledge of HIV dynamics can be assessed by referring to the prior sections of this paper. Clearly, many effects are aggregated and simplified; the core notion is that infection of Th cells is the dominant dynamic characteristic. Explicit cytotoxic and phagocytic immune effects are not modeled but are incorporated in parameter definitions; a discussion of this assumption can be found in [69]. Equations 1-4 do not account for mutation, whose effects are considered in a later section. Figure 2. Untreated response to wild-type HIV infection. Optimal Therapy for Wild-Type Infection The optimal therapeutic protocol is derived by minimizing a treatment cost function, J, that penalizes large values of HIV and infected Th cell concentrations at the end time and during the fixed time interval [to, tf], as well as excessive application of therapeutic agents during the interval. The scalar cost function takes the general form, J= tf % 1" T #x (t f )S f x(t f ) + ! [x T (t)Qx(t ) + uT (t)Ru(t)]dt & 2$ to ' tf [ ] = ( x(t f ) + ! L[ x(t), u(t)]dt Figure 1. Overview of the dynamic model for HIV-immune system interaction. to (5a) The control vector is different from that considered in [89]; its elements are concentrations of protease inhibitor (u1), fusion inhibitor (u2), Th cell enhancer (u3), and reverse transcription inhibitor (u4). The controls enter as factors that either attenuate or enhance naturally occurring ! 1 J = s f 11 x1 f 2 + s f 33 x 3 f 2 + s f 44 x 4 f 2 2 ( 1 + 2 ) tf " [q 2 2 2 ] x + q33 x 3 + q44 x 4 + rui dt 11 1 to 2 (5b) Mutation and Control of the Human Immunodeficiency Virus where x T = [ x1 x 2 x 3 x 4 ] , u = ui (i = 1 or 4), and the diagonal matrices S, Q, and R establish relative weights that penalize variations from zero of the final state and the state and control over the time interval of interest. The ! variables are squared to amplify the effects of large variations and to de-emphasize contributions of small variations. Each squared element is multiplied by a coefficient that establishes the relative importance of the factor in the treatment cost. The coefficients (sii, qii, and r) could reflect financial cost, or they could represent physiological "cost," such as toxicity or discomfort. Thus, there is a mechanism for trading one variation against the others in defining the treatment protocol, balancing speed and efficacy of treatment against implicit side effects. Methods for minimizing the cost function with respect to the control and subject to the dynamic constraint are well known; they are presented in [95] and applied to an immune response problem in [96]. A Hamiltonian is formed from the Lagrangian (the integrand of eq. 5) with the dynamic constraint adjoined by Lagrange multipliers, and the Euler-Lagrange equations (necessary conditions for optimality) are satisfied via steepest-descent numerical iteration. Further inequality constraints are applied to assure that all variables remain positive and do not exceed specified limits. The result is an optimal trajectory x*(t) in [to, tf] that is generated by the optimal control profile, u*(t), in the same time interval. An example based on protease inhibitor therapy is shown in Fig. 3. Here, the PI is assumed to have 100% efficacy. The maximum dosage is maintained for about two months, during which time the viral and infected Th cell concentrations are reduced dramatically. At the end of the period, the PI concentration is decreased toward a maintenance level that reaches a non-zero steady state (not shown). If the PI efficacy is reduced to 90%, the duration of maximum dosage is doubled and the decay rate of infection is slowed, but the trends are the same (Fig. 4). 5 RTI monotherapy is about as effective in taming the wild-type infection as is PI therapy (Fig. 5). Because the agent enters at an earlier stage of the HIV replication sequence, proviral Th cell concentration continues to grow for a few weeks after therapy has begun, then decays over the next several months. These results suggest that PI and RTI therapy would be quite effective in controlling HIV if there were no mutation. Figure 4. Effect of optimal protease inhibitor therapy with 90% efficacy. Figure 5. Effect of optimal reverse transcriptase inhibitor therapy with 100% efficacy. Figure 3. Effect of optimal protease inhibitor therapy with 100% efficacy. Although not shown, two other therapeutic effects have been calculated and are of interest. Because a fusion inhibitor (u2) impedes the replication process at an early stage, its optimal effect is similar to that of an RTI; hence, its dynamic history is similar to Fig. 5. Optimizing therapy with the cost function of eq. 5 and a notional Th cell enhancer (u3) has a counter-intuitive effect: it calls for killing healthy Th Cells (through negative enhancement)! The reason is that Th cell concentration does not appear in the cost function. Because healthy Th cells become HIV factories, the only way such a control can reduce viral and infected Th cell concentrations is to attack healthy Th cells. On further thought, this seemingly anomalous re- Mutation and Control of the Human Immunodeficiency Virus sult suggests that temporary immune system suppression could play a role in HIV treatment, as it already does for recovery from organ transplants (as suggested by Paul Shapshak, University of Miami, in personal communication). Penalizing deviation from a healthy Th cell population in the cost function gives rise to time-variations in both viral and healthy Th cell levels that require more study. Model of Host-Pathogen Interaction with Wild-Type and Mutant Infection Regrettably, mutation does occur in HIV replication, and it leads to drug-resistant strains of the virus. Thus, by definition, the new strains do not respond to treatment, but it remains to be seen if their dynamic interactions with drug-sensitive strains can be used to good effect. To examine this possibility, we examine a single mutant strain and its effects by appending three equations to our original fourth-order model: 6 decays as before (Fig. 6). The rate of growth is about the same for both strains, although the mutant strain has lower net concentration. Lowering the mutant fitness to 10% produces a similar result (Fig. 7), but the mutant concentration is lower still. Throughout the simulated period, both mutant strains grow, but the wild-type strain remains responsible for the bulk of infection. In this model, the wild-type strain does not explicitly suppress the mutant strain through competition for resources, but it does remain the dominant factor. x˙1 = "a1 x1 " a2 x1 x 2 (1" u2 ) + a3 a4 x 4 (1" a10 )(1" u1 ) a5 x˙ 2 = " a2 x1 x 2 (1" u2 ) " a6 x 2 " a2 a11 x 5 x 2 1+ x1 + x 5 # x + x3 + x4 + x6 + x7 & +a7 %1" 2 ( x 2 (1+ u3 ) a8 $ ' x˙ 3 = a2 x1 x 2 (1" u2 ) " a9 x 3 (1" u4 ) " a6 x 3 x˙ 4 = a9 x 3 (1" u4 ) " a4 x 4 x˙ 5 = a3 a4 a10 x 4 + a3 a4 x 7 " a1 x 5 " a2 a11 x 5 x 2 x˙ 6 = a2 a11 x 5 x 2 " a9 x 6 " a6 x 6 x˙ 7 = a9 x 6 " a4 x 7 Figure 6. Untreated response to wild-type and mutant infection. Mutant fitness = 90%. (6-12) ! The new state elements represent concentrations of the mutant HIV strain (x5), proviral Th cells infected by the mutant strain (x6), and Th cells productively infected by the new strain (x7). The differential equations for these three variables (eq. 10-12) are modeled after eq. 1, 3, and 4, but they do not contain direct effects of therapy. They are, however, coupled to the previous equations by a mutation rate term (with multiplier a10), and they contain a decay term representing “fitness” that is moderated by a11. The wild-type strain can be assumed to have a fitness of one (after [97]), and the mutant strain has a fitness (a11) less than one (else it would be a wild-type strain through evolution). The equation governing the production of healthy Th cells (eq. 7) also is modified to account for the new populations of virions and infected cells. For illustration, we assume that the mutation rate is 10%, double the highest suggested value in the literature, and we allow the mutant-strain fitness to vary between 10% and 90%. ! With a mutant fitness of 90% and no therapy, both HIV strains progress, and the healthy Th cell population Figure 7. Untreated response to wild-type and mutant infection. Mutant fitness = 10%. Optimal Therapy for Wild-Type Plus Mutant Infection Therapy is optimized for the revised system. The cost function is reframed to penalize the growth of the mutant strain and its infection of Th cells: J= 1 + 2 1 s f 55 x 5 f 2 + s f 66 x 6 f 2 + s f 77 x 7 f 2 2 ( ) tf " [q 2 2 2 (13) ] x + q66 x 6 + q77 x 7 + rui dt 55 5 to 2 The wild-type strain and its effects are not considered in the cost, and the control cost is the same as in eq. 5. With a mutant fitness of 90%, PI effect on the wild-type HIV strain is quite similar to the unmutated case; the wild type Mutation and Control of the Human Immunodeficiency Virus is virtually eliminated within the first few weeks through the action of maximum drug dosage. Nevertheless, the mutant strain grows; it contributes to reduction in the number of healthy Th cells, although at a lower rate than in the untreated case (Fig. 6). The optimal therapy remains at or near the full value for the simulated period. 7 the mutant strain has low fitness. “Start-stop therapy” or “drug holidays” are largely discounted by clinical trials [14, 31], although there are somewhat mixed results. Differences in experimental findings could arise from dissimilar fitness levels in actual mutant strains. There is, of course a major caveat: we have no control over the fitness of mutant strains, and there is every reason to believe that mutations at every fitness level occur. Nevertheless, the present result shows us that monotherapy can have an effect on controlling at least some proportion of the drug-resistant mutant population. Multi-drug therapy targets different mutant strains, increasing overall efficacy. If fitness, drug resistance, and progression of the disease are related, as suggested in [44, 45, 97], optimizing multi-drug strategies may reveal synergistic effects that are currently unknown, providing better control of the virus and reducing harmful side effects of treatment. Figure 8. Response of wild-type and mutant virus to optimal protease inhibitor therapy. Mutant fitness = 90%. Reducing the mutant fitness level to 70% has little effect on the optimal PI therapy history (Fig. 9), but the healthy Th cell count is maintained at high levels for the period. The wild-type HIV is controlled as before, and the mutant strain grows at a much lower rate. A further 10% reduction in mutant fitness has a dramatic effect: the mutant concentration and its effects diminish over time. The optimal dosage is smaller toward the end of the period, suggesting that the situation is in control. Thus, there is a dividing point between success and failure in controlling the drug-resistant mutant strain at a fitness level between 60% and 70%. Figure 10. Response of wild-type and mutant virus to protease inhibitor. Mutant fitness = 60%. Figure 11. Response of wild-type and mutant virus to protease inhibitor. Mutant fitness = 20%. Figure 9. Response of wild-type and mutant virus to protease inhibitor. Mutant fitness = 70%. V. REFLECTIONS AND EXTENSIONS With reduction of mutant fitness to 20%, there is significant reduction in optimal PI dosage, but the dosage is increasing toward the end of the period. This may suggest optimality of a structured reduction in therapy when The suggestions for HIV therapy that are presented here are optimal only to the extent that the mathematical models are accurate reflections of host-pathogen-pharma- Mutation and Control of the Human Immunodeficiency Virus ceutical interactions. Complex issues of pharmacokinetics have not been addressed, under the assumption that prescribed levels of therapeutic agents could be maintained in circulatory and lymphatic systems through unspecified processes. The circulatory and lymphatic systems have been treated as a single control volume, even though we know that host-pathogen interactions and residence times are quite different in the periphery and in various lymphoid tissues. Furthermore, no two individuals respond to infection and treatment in quite the same way, in large part because the rest of the body’s systems couple into HIV response. The immune system’s active response is only inferred in the model, the dynamics of CD4 cells other than Th cells have been neglected, and details of viral reservoirs remain to be determined. There may never be answers to reasonable questions about the nature of important physiological traits and the potential direct and side effects of therapy. Still, there is much room and cause for future work on optimal therapies for HIV. Some of the issues just raised have been treated in references mentioned earlier, so there is a rich foundation for further application of control-theoretic principles. References [98, 99] present an introduction to dealing with uncertainty and incompleteness in immune system models, measurements, and control action, and many other approaches to design and analysis remain to be investigated. Through the normal course of biomedical research, models will improve, allowing control theory to make significant contributions to clinical practice. 8 sity of Miami, has been the source of much information on the fundamentals of HIV infection. Undergraduate research by David Sachs, '02, and Hugh Strange, '04, provided preliminary analyses that were helpful in formulating the current study. REFERENCES 1. 2. 3. 4. 5. 6. 7. 8. CONCLUSION 9. Numerical optimization of nonlinear models can suggest improved therapies for treating HIV infection. The dynamics of infection are far more complex and varied than portrayed by the simple mathematical models used here, but the analytical results give structures and rationales for treatment that could prove foundational for future practice. In particular, the study shows that current drug classes can provide indirect control of low-fitness drugresistant HIV strains through dynamic coupling to wildtype strains and immune-system dynamics. Extension of this approach to multi-drug HIV therapy is straightforward and is the logical next step. 10. 11. ACKNOWLEDGEMENT This research was initiated under a grant from the Alfred P. Sloan Foundation. This paper was written while the author was on sabbatical leave from Princeton University as a member of the Institute for Advanced Study, Princeton, NJ. Discussions with Alan Perelson, Los Alamos National Laboratory, Arnold Levine, director of the Institute’s Center for Systems Biology, and members of the Center have been invaluable. Paul Shapshak, Univer- 12. 13. Gallo, R. C., and Montagnier, L. (1988). “AIDS in 1988,” Scientific American, 259, pp. 41-48. Prusiner, S. B. (2002). “Discovering the Cause of AIDS,” Science, 298, pp. 1726-1727. Montagnier, L. (2002). “A History of HIV Discovery,” Science, 298, pp. 1727-1728. Gallo, R. C. (2002). “The Early Years of HIV/AIDS,” Science, 298, pp. 1728-1730. Weiss, R. A. (1993). “How Does HIV Cause AIDS?” Science, 260, pp. 1273-1278. Dornadula, G., Zhang, H., VanUitert, B., Stern, J., Livornese, Jr., L., Ingerman, M. J., Witek, J., Kedanis, R. J., Natkin, J., DeSimone, J., and Pomerantz, R. J. (1999). “Residual HIV-1 RNA in Blood Plasma of Patients Taking Suppressive Highly Active Antiretroviral Therapy,” J. American Medical Association, 282 (17), pp. 1627-1632. Zhang, L., Ramratnam, B., Tenner-Racz, K., He, X., Vesanen, M., Lewin, S., Talal, A., Racz, P., Perelson, A. S., Korber, B. T., Markowitz, M., and Ho, D. D. (1999). “Quantifying Residual HIV-1 Replication in Patients Receiving Combination Antiretroviral Therapy,” New England J. Medicine, 340 (21), pp. 1605-1613. Havlir, D. V., Hellmann, N. S., Petropoulos, C. J., Whitcomb, J. M., Collier, A. C., Hirsch, M. S., Tebas, P., Sommadossi, J.-P., and Richman, D. D. (2000). “Drug Susceptibility in HIV Infection After Viral Rebound in Patients Receiving Indinavir-Containing Regimens,” J. American Medical Association, 283 (2), pp. 229-234. Furtado, M. R., Callaway, D. S., Phair, J. P., Kunstman, K. J., Stanton, J. L., Macken, C. A., Perelson, A. S., and Wolinsky, S. M. (1999). “Persistence of HIV-1 Transcription in Peripheral-Blood Mononuclear Cells in Patients Receiving Potent Antiretroviral Therapy,” New England J. Medicine, 340 (21), pp. 1614-1622. Ramratnam, B., Mittler, J. E., Zhang, L., Boden, D., Hurley, A., Fang, F., Macken, C. A., Perelson, A. S., Markowitz, M., and Ho, D. D. (2000). “The Decay of the Latent Reservoir of Replication-Competent HIV-1 is Inversely Correlated with the Extent of Residual Viral Replication during Prolonged Anti-Retroviral Therapy,” Nature Medicine, 6 (1), pp. 82-85. Sharkey, M. E., Teo, I., Greenough, T., Sharova, N., Luzuriaga, K., Sullivan, J. L., Bucy, R. P., Kostrikis, L. G., Haase, A., Veryard, C., Davaro, R. E., Cheeseman, S. H., Daly, J. S., Bova, C., Ellison, R. T., III, Mady, B., Lai, K. K., Moyle, G. Nelson, M., Gazzard, B., Shaunak, S., and Stevenson, M. (2000). “Persistence of Episomal HIV-1 Infection Intermediates in Patients on Highly Active AntiRetroviral Therapy,” Nature Medicine, 6 (1), pp. 76-81. Fraser, C., Ferguson, N. M., and Anderson, R. M. (2001). “Quantification of Intrinsic Residual Viral Replication in Treated HIV-Infected Patients,” Proc. National Academy of Science, 98 (26), pp. 15167-15172. Di Mascio, M., Ribeiro, R. M., Markowitz, M., Ho, D. D., and Perelson, A. S. (2004). “Modeling the Long-Term Mutation and Control of the Human Immunodeficiency Virus 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. Control of Viremia in HIV-1 Infected Patients Treated with Antiretroviral Therapy,” Mathematical Biosciences, 188, pp. 47-62. Bartlett, J. G., and Moore, R. D. (1998). “Improving HIV Therapy,” Scientific American, 279, pp. 84-87, 89, 91-93. Salk, J., Bretscher, P. A., Salk, P. L., Clerici, M., and Shearer, G. M. (1993). “A Strategy for Prophylactic Vaccination Against HIV,” Science, 260, pp. 1270-1272. Redfield, R. R., and Burke, D. S. (1988). “HIV Infection: The Clinical Picture,” Scientific American, 259, pp. 90-98. Gougeon, M.-L., and Montagnier, L. (1993). “Apoptosis in AIDS,” Science, 260, pp. 1269-1270. McCune, J. M. (2001). “The Dynamics of CD4+ T-Cell Depletion in HIV Disease,” Nature, 410, pp. 974-979. Janeway, C. A., Travers, P., Walport, M., and Shlomchik, M. (2001). Immunobiology, Garland, London. Douek, D. C., Brenchley, J. M., Betts, M. R., Ambrozak, D. R., Hill, B. J., Okamoto, Y., Casazza, J. P., Kuruppu, J., Kunstman, K., Wollinsky, S., Grossman, Z., Dybul, M., Oxenius, A., Price, D. A., Connors, M. and Koup, R. A. (2002). “HIV Preferentially Infects HIV-Specific CD4+ T Cells,” Nature, 417, pp. 95-98. Schacker, T., Little, S., Connick, E., Gebhard, K., Zhang, Z.-Q., Krieger, J., Pryor, J., Havlir, D., Wong, J. K., Schooley, R. T., Richman, D., Corey, L., and Haase, A. T. (2001). “Productive Infection of T Cells in Lymphoid Tissues during Primary and Early Human Immunodeficiency Virus Infection,” J. Infectious Diseases, 183, pp. 555-562. Klebanoff, S. J., and Coombs, R. W. (1997). “Virucidal Effect of Stimulated Eosinophils on Human Immunodeficiency Virus Type 1,” AIDS Res. Hum. Retroviruses, 12 (1), pp. 25-29. Shapshak, P., Fujimura, R. K., Srivastava, A., and Goodkin, K. (2000). “Dementia and the Neurovirulence of HIV1,” CNS Spectrums, 5 (4), pp. 31-42. Garden, G. A. (2002). “Microglia in Human Immunodeficiency Virus-Associated Neurodegeneration,” Glia, 40 (2), pp. 240-251. Orenstein, J. A., Fox, C., and Wahl, S. M. (1997). “Macrophages as a Source of HIV During Opportunistic Infections,” Science, 276, pp. 1857-1861. Ponath, P. D., Qin, S., Post, T. W., Wang, J., Wu, L., Gerard, N. P., Newman, W., Gerard, C., and Mackay, C. R. (1996). “Molecular Cloning and Characterization of a Human Eotaxin Receptor Expressed Selectively on Eosinophils,” J. Experimental Medicine, 183, pp. 2437-2448. de Paulis, A., De Palma, R., Di Gioia, L., Carfora, M., Prevete, N., Tosi, G., Accolla, R. S., and Marone, G. (2000). “Tat Protein Is an HIV-1-Encoded β-Chemokine Homolog That Promotes Migration and Up-Regulates CCR3 Expression on Human FceRI+ Cells,” J. Immunology, 165, pp. 7171-7179. Lempicki, R. A., Kovacs, J. A., Baseler, M. W., Adelsberger, J. W., Dewar, R. L., Natarajan, V., Bosche, M. C., Metcalf, J. A., Stevens, R. A., Lambert, L. A., Alvord, W. G., Polis, M. A., Davey, R. T., Dimitrov, D. S., and Lane, H. C. (2000). “Impact of HIV-1 Infection and Highly Active Antriretroviral Therapy on the Kinetics of CD4+ and CD8+ T Cell Turnover in HIV-Infected Patients,” Proc. National Academy of Science, 97 (25), pp. 13778-13783. Papagno, L., Spina, C. A., Marchant, A., Salio, M., Rufer, N., Little, S., Dong, T., Chesney, G., Waters, A., Easterbrook, P., Dunbar, P. R., Shepherd, D., Cerundolo, V., Emery, V., Griffiths, P., Conlon, C., McMichael, A. J., Rich- 9 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. man, D. D., Rowland-Jones, S. L., and Appay, V. (2004). “Immune Activation and CD8+ T-Cell Differentiation Towards Senescence in HIV-1 Infection,” PLoS Biology, 2 (2), pp. 173-185. McMichael, A. J., and Rowland-Jones, S. L. (2001). “Cellular Immune Responses to HIV,” Nature, 410, pp. 980-987. McMichael, A. J., and Rowland-Jones, S. L. (2002). “Bad News for Start-Stop Therapy?” Nature, 420, pp. 371-372. Nowak, M. A., May, R. M., Phillips, R. E., Rowland-Jones, S., Lalloo, D. G., McAdam, S., Klenerman, P., Köppe, Sigmund, K., Bangham, C. R. M., and McMichael, A. J. (1995). “Antigenic Oscillations and Shifting Immunodominance in HIV-1 Infections,” Nature, 375, pp. 606-611. Grant, R. M., Hecht, F. M., Warmerdam, M., Liu, L., Liegler, T., Petropoulos, C. J., Hellman, N. S., Chesney, M., Busch, M. P., and Kahn, J. O. (2002). “Time Trends in Primary HIV-1 Drug Resistance Among Recently Infected Persons,” J. American Medical Association, 288 (2), pp. 181-188. Douek, D. C., Picker, L. J., and Koup, R. A. (2003). “T Cell Dynamics in HIV-1 Infection,” Annual Review of Immunology, 21, pp. 265-304. Ho, D. D. (1995). “Time to Hit HIV, Hard and Early,” New England J. Medicine, 333, pp. 450-451. Bonhoeffer, S., May, R. M., Shaw, G. M., and Nowak, M. A. (1997). “Virus Dynamics and Drug Therapy,” Proc. National Academy of Science, 94, pp. 6971-6976. Karlsson, A. C., Birk, M., Lindbäck, S., Gaines, H., Mittler, J. E., and Sönnerberg, A. (2001). “Initiation of Therapy during Primary HIV Type 1 Infection Results in a Continuous Decay of Proviral DNA and a Highly Restricted Viral Evolution,” AIDS Research and Human Retroviruses, 17 (5), pp. 409-416. Wang, C., Vlahov, D., Galai, N., Bareta, J., Strathdee, S. A., Nelson, K. E., and Sterling, T. R. (2004). “Mortality in HIV-Seropositive versus –Seronegative Persons in the Era of Highly Active Antiretroviral Therapy: Implications for When to Initiate Therapy,” J. Infectious Diseases, 190, pp. 1046-1054. Richman, D. D. (2001). “HIV Chemotherapy,” Nature, 410, pp. 995-1001. Hoffman, C., and Kamps, B. (2005). “HIV Drug Resistance 2005” HIV Medicine, http://www.hivmedicine.com/textbook/rt.htm . Batsis, J. A. (2000). “Clinical Pharmacology of Protease Inhibitors in HIV Infection,” Trinity Student Medical Journal, 1, pp. 60-65. Watson, J. D., Baker, T. A., Bell, S. P., Gann, A., Levine, M., and Losick, R. (2004). Molecular Biology of the Gene, Pearson/Benjamin Cummings, San Francisco. Eigen, M., and Bierbricher, C. K. (1988). “Sequence Space and Quasispecies Distribution,” in DNA Genetics, Volume 3: Variability of RNA Genomes, E. Domingo, J. J. Holland, and P. Ahlquist, ed., CRC Press, Boca Raton. Quiñones-Mateu, M. E., and Arts, E. J. (2001). “HIV-1 Fitness: Implications for Drug Resistance, Disease Progression, and Global Epidemic Evolution,” HIV Sequence Compendium, Los Alamos National Laboratory, Los Alamos, NM, pp. 134-170. Quiñones-Mateu, M. E., Ball, S. C., Marozsan, A. J., Torre, V. S., Albright, J. L., Vanham, G., van der Groen, G., Colebunders, R. L., and Arts, E. J. (2000). “A Dual Infection/Competition Assay Shows a Correlation Between Ex Mutation and Control of the Human Immunodeficiency Virus 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. Vivo Human Immunodeficiency Virus Type 1 Fitness and Disease Progression,” J. Virology, 74 (19), pp. 9222-9233. Joly, V., and Yeni, P. (1999). “Non Nucleoside Reverse Transcriptase Inhibitors,” AIDS Reviews, 1 (1), pp. 37-44. Ciancio, B. C., Trotta, M. P., Lorenzini, P., Forbici, F., Visco-Comandini, U., Gori, C., Bonfigli, S., Bellohi, M. C., Sette, P., D’Arrigo, R., Tozzi, V., Zaccarelli, M., Boumis, E., Narciso, P., Perno, C. F., and Antinoni, A. (2003). “The Effect of Number of Mutations and of Drug-Class Sparing on Virological Response to Salvage Genotype-Guided Antiretroviral Therapy,” Antiviral Therapy, 8 (6), pp. 611616. Kilby, J. M., and Eron, J. J. (2003). “Novel Therapies Based on Mechanisms of HIV-1 Cell Entry,” New England J. Medicine, 348 (22), pp. 2228-2238. Witvrouw, M., Van Maele, B., Vercammen, J., Hantson, A., Engelborghs, Y., De Clerq, E., and Debyser, Z.(2004). “Novel Inhibitors of HIV-1 Integration,” Curr. Drug Metab., 5 (4), pp. 291-304. Li, F., Goila-Gaur, R., Salzwedel, K., Kilgore, N. R., Reddick, M., Matallana, C., Castillo, A., Zoumplis, D., Martin, D. E., Orenstein, J. M., Allaway, G. P., Freed, E. O., and Wild, C. T. (2003). “PA-457: A Potent HIV Inhibitor that Disrupts Core Condensation by Targeting a Late Step in Gag Processing,” Proc. National Academy of Science, 100 (23), pp. 13555-13560. Lum, J. L., Schnepple, D. J., Nie, Z., Sanchez-Dardon, J., Mbisa, G. L., Mihowich, J., Hawley, N., Narayan, S., Kim, J. E., Lynch, D. H., and Badley, A. D. (2004). “Differential Effects of Interleukin-7 and Interleukin-15 on NK Cell Anti-Human Immunodeficiency Virus Activity,” J. Virology, 78 (11), pp. 6033-6042. Casado, J. L., Moreno, A., Sabido, R., Marti-Belda, P., Antela, A., Drona, F., Perez-Elias, M. J., and Moreno, S. (2003). “Individualizing Salvage Regimens: The Inhibitory Quotient (Ctrough/IC50) as Predictor of Virological Response,” AIDS, 17(2), pp. 262-264. Evans, W. E., McLeod, H. L. (2003). “Pharmacogenomics – Drug Disposition, Drug Targets, and Side Effects,” New England J. Medicine, 348, pp. 538-549. Gallant, J. E., Gerondelis, P. Z., Wainberg, M. A., Shulman, N. S., Haubrich, R. H., St. Clair, M., Lanier, E. R., Hellman, N. S., and Richman, D. D. (2003). “Nucleoside and Nucleotide Analogue Reverse Transcriptase Inhibitors: A Clinical Review ofAntiretroviral Resistance,” Antiviral Therapy, 8, pp. 489-506. Burton, D. R., Desrosiers, R. C., Doms, R. W., Koff, W. C., Kwong, P. D., Moore, J. P., Nabel, G. J., Sodroski, J., Wilson, I. A., and Wyatt, R. T. (2004). “HIV Vaccine Design and the Neutralizing Antibody Problem,” Nature Immunology, 5 (3), pp. 233-236. Capdevila, G., “AIDS Vaccine Elusive – Even for Patents,” http://www.aegis.com/news/ips/2005/IP050207.html, Inter Press Service, Feb. 8. 2005. Covert, D. J., and Kirschner, D. (2000) “Revisiting Early Models of the Host-Pathogen Interactions in HIV Infection,” Comments on Theoretical Biology, 5 (6), pp. 383411. Cooper, L. (1986). “Theory of an Immune System Retrovirus,” Proc. National Academy of Science, 83, pp. 91599163. Perelson, A. S. (1989). Modeling the Interaction of the Immune System with HIV. in Mathematical and Statistical Approaches to AIDS Epidemiology (C. Castillo-Chavez, ed.), Springer-Verlag, New York, pp. 350-370. 10 60. Kirschner, D. (1996). “Using Mathematics to Understand HIV Dynamics,” Notices of the AMS, 43 (2), pp. 191-202. 61. de Jong, M. D., Veenstra, J., Stillianakis, N. I., Schuurman, R., Lange, J. M. A., de Boer, R. J., and Boucher, C. A. B. (1996). “Host-Parasite Dynamics and Outgrowth of Virus Containing a Single K70R Amino Acid Change in Reverse Transcriptase are Responsible for the Loss of Human Immunodeficiency Virus Type 1 RNA Load Suppression by Zidovudine,” Proc. National Academy of Science, 93, pp. 5501-5506. 62. de Boer, R. J., and Boucher, C. A. B. (1996). “Anti-CD4 Therapy for AIDS Suggested by Mathematical Models,” Proc.: Biological Sciences, The Royal Society, 263 (1372), pp. 899-905. 63. Kirschner, D. E., and Webb, G. F. (1997). “A Mathematical Model of Combined Drug Therapy of HIV Infection,” J. Theoretical Medicine, 1, pp. 25-34. 64. Stillianakis, N. I., Boucher, C. A. B., de Jong, M. D., van Leeuven, R., Schuurman, R., and de Boer, R. J. (1997). “Clinical Data Sets of Human Immunodeficiency Virus Type 1 Reverse Transcriptase-Resistant Mutants Explained by a Mathematical Model,” J. Virology, 71 (1), pp. 161168. 65. Bonhoeffer, S., and Nowak, M. A. (1997). “Pre-Existence and Emergence of Drug Resistance in HIV-1 Infection,” Proc.: Biological Sciences, The Royal Society, 264 (1382), pp. 631-637. 66. Nowak, M. A., Bonhoeffer, S., Shaw, G. M., and May, R. M. (1997). “Anti-Viral Drug Treatment: Dynamics of Resistance in Free Virus and Infected Cell Population,” J. Theoretical Biology, 184, pp. 203-217. 67. Kirschner, D. E., and Webb, G. F. (1998). “Immunotherapy of HIV-1 Infection,” J. Biological Systems, 6 (1), pp. 7183. 68. Wein, L. M., D’Amato, R. M., and Perelson, A. S. (1998). “Mathematical Analysis of Antiretroviral Therapy Aimed at HIV-1 Eradication or Maintenance of Low Viral Loads,” J. Theoretical Biology, 192, pp. 81-98. 69. de Boer R. J., and Perelson, A. S. (1998). “Target Cell Limited and Immune Control Models of HIV Infection: A Comparison,” J. Theoretical Biology, 190, pp. 201-214. 70. Perelson, A. S., and Nelson, P. W. (1999). “Mathematical Analysis of HIV-1 Dynamics in Vivo,” SIAM Review, 41 (1), pp. 3-44. 71. Wodarz, D., Lloyd, A. L., Jansen, V. A. A., and Nowak, M. A. (1999). “Dynamics of Macrophage and T Cell Infection by HIV,” J. Theoretical Biology, 196, pp. 101-113. 72. Wodarz, D., and Nowak, M. A. (1999). “Specific Therapy Regimes Could Lead to Long-Term Immunological Control of HIV,” Proc. National Academy of Sciences, 96 (25), pp. 14464-14469. 73. Stafford, M. A., Cao., Y. C., Ho, D. D., Corey, L., and Perelson, A. S. (2000). “Modeling Plasma Virus Concentration and CD4+ T Cell Kinetics during Primary HIV Infection,” J. Theoretical Biology, 203, pp. 285-301. 74. Kaufmann, G. R., Zaunders, J., Murray, J. Kelleher, A. D., Lewin, S. R., Solomon, A., Smith, D., and Cooper, D. A. (2001). “Relative Significance of Different Pathways of Immune Reconstitution in HIV Type 1 Infection as Estimated by Mathematical Modeling,” AIDS Research and Human Retroviruses, 17 (2), pp. 147-159. 75. Wodarz, D., and Jansen, V. A. A. (2001). “The Role of T Cell Help for Anti-viral CTL Responses,” J. Theoretical Biology, 211, pp. 419-432. Mutation and Control of the Human Immunodeficiency Virus 76. Callaway, D. S., and Perelson, A. S. (2002). “HIV-1 Infection and Low Steady State Viral Loads,” Bul. Mathematical Biology, 64, pp. 29-64. 77. Bajaria, S. H., Webb, G., Cloyd, M., and Kirschner, D. (2002). “Dynamics of Naïve and Memory CD4+ T Lymphocytes in HIV-1 Disease Progression,” J. Acquired Immune Deficiency Syndromes, 30 (1), pp. 41-58. 78. Nelson, P. W., and Perelson, A. S. (2002). “Mathematical Analysis of Delay Differential Equation Models of HIV-1 Infection,” Mathematical Biosciences, 179, pp. 73-94. 79. Altes, H. K., Wodarz, D., and Jansen, V. A. A. (2002). “The Dual Role of CD4 T Helper Cells in the Infection Dynamics of HIV and Their Importance for Vaccination,” J. Theoretical Biology, 214, pp. 633-646. 80. Culshaw, R. V., Ruan, S., and Webb, G. (2003). “A Mathematical Model of Cell-to-Cell Spread of HIV-1 that Includes a Time Delay,” J. Mathematical Biology, 46, pp. 425-444. 81. Bajaria, S. H., Webb, G., and Kirschner, D. E. (2004). “Predicting Differential Responses to Structured Treatment Interruptions During HAART,” Bul. Mathematical Biology, 66 (5), pp. 1093-1118, 82. Dixit, N., and Perelson, A. S. (2004). “Complex Patterns of Viral Load Decay under Antiretroviral Therapy; Influence of Pharmacokinetics and Intracellular Delay,” J. Theoretical Biology, 226, pp. 95-109. 83. Iwasa, Y., Michor, F., and Nowak, M. (2004). “Some Basic Properties of Immune Selection,” J. Theoretical Biology, 229, pp. 179-188. 84. Kirschner, D. (1999). “Dynamics of Co-Infection with M. Tuberculosis and HIV-1,” Theo. Population Biology, 55, pp. 94-109. 85. Kirschner, D., Mehr, R., and Perelson, A. (1998). “Role of the Thymus in Pediatric HIV-1 Infection,” J. Acq. Imm. Def. Synd. And Hum. Retro., 18 (2), pp. 95-109. 86. Ye, P., and Kirschner, D. E. (2002). “Reevaluation of T Cell Receptor Excision Circles as a Measure of Recent Thymic Emigrants,” J. Immunology, 168 (10), pp. 49684979. 87. Ye,. P., Kourtis, A., and Kirschner, D. (2002). “The Effects of Different HIV Type 1 Strains on Human Thymic Function,” AIDS Res. And Hum. Retr., 18(7), pp. 1239-1251. 88. Ye,. P., Kourtis, A., and Kirschner, D. (2003). “Reconstitution of Thymic Function in HIV-1 Patients Treated with Highly Active Antiretroviral Therapy,” Clinical Immunology, 106, pp. 95-105. 89. Kirschner, D., Lenhart, S., and Serbin, S. (1997). “Optimal Control of the Chemotherapy of HIV,” J. Mathematical Biology, 35, pp. 775-792. 90. Wein, L. M., Zenios, S. A., and Nowak, M. A. (1997). “Dynamic Multidrug Therapies for HIV: A Control Theoretic Approach,” J. Theoretical Biology, 185, pp. 15-29. 91. Alvarez-Ramirez, J., Meraz, M., and Velasco-Hernández, J. X. (2000). “A Feedback Control Strategy for Chemotherapy of HIV,” Intl. J. Bifurcations and Chaos, 10 (9), pp. 2207-2219. 92. Velasco-Hernández, J. X., Garcia, J. A., and Kirschner, D. E. (2001). “Remarks on Modeling Host-Viral Dynamics and Treatment,” in Vol.125: Mathematical Approaches for Emerging and Reemerging Infectious Diseases Part 1: An Introduction to Models, Methods, and Theory (C. CastillioChavez, S. Blower, P. van den Driessche, D. Kirschner, and A.-A. Yakubu, ed.), pp. 338-411. 11 93. Brandt, M. E., and Chen, G. (2001). “Feedback Control of a Biodynamical Model of HIV-1”, IEEE Trans. Biomedical Engineering, 48 (7), pp. 754-759. 94. Culshaw, R. L., Ruan, S., and Spiteri, R. J. (2004). “Optimal HIV Treatment by Maximising Immune Response,” J. Mathematical Biology, 48, pp. 542-564. 95. Stengel, R. F. (1994). Optimal Control and Estimation, Dover Publications, New York. 96. Stengel, R. F., Ghigliazza, R., Kulkarni, N., and Laplace, O. (2002). "Optimal Control of Innate Immune Response," Optimal Control Applications & Methods, 23, pp. 91-104. 97. Nowak, M. A., and May, R. M. (2000). Virus Dynamics, Oxford University Press, Oxford. 98. Stengel, R. F., Ghigliazza, R., and Kulkarni, N. (2002). "Optimal Enhancement of Immune Response," Bioinformatics, 18 (9), pp. 1227-1235. 99. Stengel, R. F., and Ghigliazza, R. (2004). "Stochastic Optimal Enhancement of Immune Response," Mathematical Biosciences, 191, pp. 123-142.