Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Equilibrium chemistry wikipedia , lookup
Nitrogen-vacancy center wikipedia , lookup
Mössbauer spectroscopy wikipedia , lookup
Electron scattering wikipedia , lookup
Degenerate matter wikipedia , lookup
Magnetic circular dichroism wikipedia , lookup
Photoelectric effect wikipedia , lookup
Heat transfer physics wikipedia , lookup
X-ray photoelectron spectroscopy wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Hartree–Fock method wikipedia , lookup
Metastable inner-shell molecular state wikipedia , lookup
Transition state theory wikipedia , lookup
Marcus theory wikipedia , lookup
X-ray fluorescence wikipedia , lookup
Rutherford backscattering spectrometry wikipedia , lookup
Chemical bond wikipedia , lookup
Photoredox catalysis wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
Atomic orbital wikipedia , lookup
Stability constants of complexes wikipedia , lookup
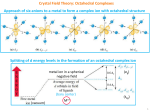
Chem 310 Lectures by: Dr. Muhammad D. Bala Office: Block H, 3-64 Contacts: phone Î extn. 2616; e-mail Î [email protected] Recommended texts: • Shriver and Atkins: Inorg. Chem., 4th Ed. • Cotton, Wilkinson and Gaus: Basic Inorg. Chem., 3rd Ed. • David Nicholls: Complexes and 1st row transition elements In this part of the course we have about 6 weeks to cover the following topics: 1.Bonding: VBT, CFT, LFT and MOT 2.Magnetic and electronic properties of TM oxides 3.Reactivities of TM complexes The Valence-Bond Theory Covalent bonds by sharing pairs of electrons was first proposed by G. N. Lewis in 1902. It was not until 1927, however, that Walter Heitler and Fritz London showed how the sharing of pairs of electrons holds a covalent molecule together. The Heitler-London model of covalent bonds was the basis of the valence-bond theory. The last major step in the evolution of this theory was the suggestion by Linus Pauling that atomic orbitals mix to form hybrid orbitals, such as the sp, sp2, sp3, dsp3, and d2sp3 orbitals. The Valence-Bond Theory It is easy to apply the valence-bond theory to some coordination complexes, such as the Co3+ complexes below. d2sp3- inner sphere complex Î low spin complex sp3d2- outer sphere complex Î high spin complex Note: Such a situation will not arise for for d1, d2 and d3 ion configuration. Deficiencies of VB approach to bonding • Assumes that all d orbitals in a complex are equal in energy. • The arbitrary use of 3d and 4d orbitals for bonding Î energy differential ignored. • The theory is unable to adequately explain electronic and magnetic properties of complexes. •VBT is widely used in organic and main group element chemistry. •In TM metal chemistry VBT is superseded by the Crystal Field Theory (CFT). •In combination with MOT it is often referred to as the Ligand Field Theory (LFT). The Crystal-Field Theory Crystal Field Theory is based on the idea that a purely electrostatic interaction exists between the central metal ion and the ligands. Covalent bonding is ignored. Crystal field theory was developed by considering two compounds: manganese(II) oxide, MnO Î octahedral geometry, copper(I) chloride, CuCl Î tetrahedral geometry. Octahedral complexes - Each Mn2+ ion in manganese(II) oxide is surrounded by six O2- ions arranged toward the corners of an octahedron. What happens to the energies of the 4s and 4p orbitals on an Mn2+ ion? z y - Mn+ - - - - Let's assume that the six O2- ions that surround each Mn2+ ion define an XYZ coordinate system. Two of the 3d orbitals (dx2-y2 and dz2) on the Mn2+ ion point directly toward the six O2- ions. The other three orbitals (dxy, dxz, and dyz) lie between the O2- ions. Therefore, the five 3d orbitals on the Mn2+ ion are no longer degenerate, i.e. no longer equal, or -of the same energy. x Octahedral Complexes z y - - •The ligands approach the metal along x, y, z. • e-s in d orbital are repulsed by –vely charged ligands Î increase in potential energy. - Mn+ - - - •Degree of e-static repulsion depends on the orientation of the d orbitals. • d orbitals (dz2 and dx2-y2) with lobes directed at the ligands experience more repulsion Î placed at higher energy eg oebitals. • d orbitals (dxy, dxz and dyx) with lobes in-between the ligands experience less repulsion Î placed at lower energy t2g orbitals. x Octahedral Complexes The s-orbital of the metal is spherically symmetric. The three p-orbitals lie along the xyz axes, Î point directly towards the ligands Î Therefore they all remain degenerate. Energy of the s- and p-orbitals is raised due to the increased repulsion between the negative point charges representing the ligands and the negative charge of the electrons orbital Î orbitals are raised in energy. Electron-density distribution in the d orbitals E z eg subset y x x dx2-y2 y t2g subset dz2 z y x x dxy z dxz dyz The Crystal-field parameter (Δ or 10 Dq) • The Crystal Field splitting parameter has been used to correlate a wide range of properties of first-row transition metal complexes. • This includes structure, electronic spectra and magnetic properties. • It is perhaps the single most useful parameter in understanding coordination chemistry. eg barycentre +6Dq 3/5Δ ο t2 2/5Δ τ Δο 3/5Δ τ -4Dq 2/5Δο 4/9 Δο e t2g octahedral field free ion tetrahedral field The Crystal-field parameter (Δ or 10 Dq) eg • The Crystal Field splitting parameter varies systematically with the type of ligand. 3 /5 Δ ο t2 2 /5 Δ τ Δο 3 /5 Δ τ 2 /5 Δ ο • E. g. in complexes of [CoX(NH3)5]n+ with X = I-, Br-, Cl-, H2O and NH3 the colours range from purple (for X = I-) thru to pink (for Cl-) to yellow (with NH3). •The same order was observed regardless of the metal ion. •Based on these observations Ryutaro Tsuchida proposed the spectrochemical series for ligands. 4 /9 Δ ο e t2 g o cta h ed ral field free io n tetra h ed ra l fie ld The Spectrochemical series I- < Br- < SCN- ~ Cl- < F- < OH- ~ ONO- < C2O42- < H2O < NCS- < EDTA4- < NH3 ~ pyr ~ en < bipy < phen < CN- ~ CO weak-field ligand. E.g. Cl- Î Low energy transition Î Δ = small strong-field ligand. E.g. CO Î High energy transition Î Δ = LARGE From a purely ionic basis we would expect CO < H2O < C2O42- < EDTA4-. That this is not the case is a reflection of covalent interactions: ignored by CFT Î Limitation of the CFT. CFT cannot account for the spectrochemical series. Why the spectrochemical series? Because the magnitude of the splitting energy Δ also determines the colours and electronic spectra of the TM complex. The Spectrochemical series The Spectrochemical series The splitting of d orbitals in the crystal field model depends on: • the geometry of the complex Î octahedral, tetrahedral, sq. planar. • the nature of the TM ion Î its group and period. • the charge on the ion. • the ligands that surround the metal. Pt 4+ > Ir 3+ > Rh 3+ Strong-field ions > Co 3+ > Cr 3+ > Fe 3+ > Fe 2+ > Co 2+ > Ni 2+ > Weak-field ions Mn 2+ Electron configuration of TM complexes Degenerate orbitals are filled according to Hund's rules: • One electron is added to each of the degenerate orbitals in a subshell before a second electron is added to any orbital in the subshell Î lowest energy subshell filled in first. • Electrons are added to a subshell with the same value of the spin quantum number until each orbital in the subshell has at least one electron Î least electrostatic repulsion. • Order of filling d-orbitals depend both on Δ and the pairing energy, P: • If Δ > P Î Δ is large, strong field ligand Îe-s pair up in the lower energy subshell first, e.g. t2g for octahedral CF Î Low spin complex Î strong field Î inner sphere. • If Δ < P Î Δ is small, weak field ligand Îe-s spread out among all d-orbitals before any pairing, e.g. t2g and eg for octahedral CF Î High-spin complex Î weak field Î outer sphere. Octahedral electron configuration of octahedral TM complexes •Îfor d1, d2, d3 and d8, d9, d10 Î the ligand type has same effect on the d-orbital electron configuration. •Î for CFSE calculation, strong or weak field ligand will be same. d1 t2g1eg0 d8 t2g6eg2 d2 t2g2eg0 d9 t2g6eg3 d3 t2g3eg0 d10 t2g6eg4 Δ < P and Δ > P Octahedral electron configuration of octahedral TM complexes d4 t2g3eg1 t2g4eg0 d5 t2g3eg2 t2g5eg0 d6 t2g4eg2 t2g6eg0 d7 t2g5eg2 t2g6eg1 Δ < P High spin Δ > P Low spin Crystal–Field Stabilisation Energy (CFSE) CFT predicts stabilisation for some electron configurations in the d orbitals. For an octahedral complex with d orbital configuration t2gxegy, with respect to the barycentre: An electron in the more stable t2g subset is treated as contributing a stabilisation of 0.4Δo OR 4Dq. An electron in the higher energy eg subset contributes to a destabilisation of 0.6Δo OR 6Dq. Therefore, the CFSE = (0.4x – 0.6y)Δo - P (P = pairing energy) OR CFSE = (4x - 6y)Dq - P (P = pairing energy) Crystal–Field Stabilisation Energy (CFSE) Q. Explain why the Co(NH3)63+ ion is a diamagnetic, low-spin complex, whereas the CoF63- ion is a paramagnetic, high-spin complex. Give the electron configuration and calculate the CFSE of each complex in terms of Δ. Answer: Co = [Ar] 3d7 4s2 Î Co3+ = [Ar] 3d6 Co(NH3)63+ = strong field ligand, Δ > P Î pairing of electrons Î t2g6 electron configuration. CoF63- = weak field ligand, Δ < P Î spreading of electrons Î t2g4 eg2 CFSE = (0.4x – 0.6y)Δo 1. Co(NH3)63+ Î x = 6 and y=0 Î CFSE = 2.4 Δ. 2. CoF63- Î x = 4 and y = 2 Î CFSE = 0.4Δ. Crystal–Field Stabilisation Energy (CFSE) Q. Determine which of the following are more likely to be high spin complexes: (1) [Fe(CN)6]3(2) [FeF6]3(3) [Co(H2O)6]+3 •(4) [Co(CN)6]-3 •(5) [Co(NH3)6]+3 •(6) [Co(en)3]+3 Solution: Compare the ligands on the spectrochemical series. Since we want a high spin complex, we want weak field ligands. The weaker field ligands in the above are H2O and F-, so complexes 2 and 3 are more likely to be high spin. (The cyanide complexes are least likely.) Crystal–Field Stabilisation Energy (CFSE) Q1. Determine the CFSE if Dq = 2100 cm-1 for the following configurations: a) d4 high spin. b) d4 low spin, assume the pairing energy P = 28,000 cm-1. Q2. Given a Dq value of 1040 and 3140 cm-1 for high spin and low spin d6 ion respectively, determine the CFSE in the following configurations if the pairing energy P = 17,600 cm-1: a ) weak field b) strong field Q1. a) d4 high spin. Configuration = t2g3; eg1. Therefore from CFSE = (-4x +6y)Dq + P CFSE = -4 x 3 + 6 = -6Dq = -12,600cm-1 ; remember for high spin up to d5 configuration P = 0 b) d4 low spin, P = 28,000 cm-1. Configuration = t2g4 Î CFSE = -4 x 4 = -16Dq + P = -5600 cm-1 Q2. a ) d6 in a weak field Configuration = t2g4; eg2. Therefore from CFSE = (-4x +6y)Dq +dP CFSE = -4 x 4 + 6 x 2 + P = -4Dq + P = 13,440cm-1 b) d6 in a strong field Configuration = t2g6. Therefore from CFSE = (-4x)Dq + 3P CFSE = -4 x 6 = -24Dq + 3P = -22,560cm-1 Tetrahedral complexes • In the tetrahedral case the electrons go to the lower energy doubly degenerate e orbital first. • The upper triply degenerate t2 orbital is filled in afterwards. • The subscripts g (gerund, german for even) and u (ungerund, german for uneven) are missing for the tetrahedral geometry. • g and u refer to inversion about a centre of symmetry which is absent for the tetrahedron. Tetrahedral complexes Note: For tetrahedral complexes, those orbitals which point towards the edges (dxy, dyz and dxz) Î e subset are raised to energy higher than those which point towards the faces Î t2 subset. That is, the exact opposite of the octahedral crystal field. A tetrahedral complex has fewer ligands, only 4 to be exact. The orbitals point in-between ligands rather than at the ligands Î lower repulsion. Due to the reasons above, the magnitude of the tetrahedral CF splitting is smaller Î Δt = 4/9 Δo Because Δt < Δo it is energetically favourable to spread out electrons Î all tetrahedral complexes are high-spin. Tetrahedral complexes Also for low spin complexes to exist Δt > P, hence low spin tetrahedral complexes of any TM configuration are very rare. Some factors that favour formation of tetrahedral complexes: Bulky ligands: inter-ligand repulsion exacerbated in an arrangement Î Steric factors octahedral Weak field ligands: Many TM halides are tetrahedral Electronic configuration of metal ion favours Δ = zero (d0, d5 and d10) or low values of OSPE Î d1, d6 and to a lesser extent d2, d7 configurations. Weak field ions: Central metal in low oxidation state Î Δ is low. Octahedral Vs Tetrahedral Summary: Field preference 1.2 Δtet = 4/9 Δoct. octahedral 1.0 Octahedral Î d3 ; d8 then d4 and d9 ) 0.8 Tetrahedral (?) Î d2; d7 d1; d6 and No influence Î d0; d5 (high spin); and d10 Octahedral Site Preference Energies, is defined as: CFSE (units of o OSPE tetrahedral 0.6 0.4 0.2 0 OSPE = CFSE (oct) CFSE (tet) OSPE 1 2 3 6 7 4 5 Number of d electrons 8 9 10 Tetragonally distorted complexes: the Jahn-Teller Effect The Jahn-Teller Theorem was published in 1937 and states: “Any non-linear molecular system in a degenerate electronic state will be unstable and will undergo distortion to form a system of lower symmetry and lower energy thereby removing the degeneracy” In simple terms it means that no nonlinear molecule can be stable in a degenerate electronic state. The molecules must become distorted to remove the degeneracy In an octahedral crystal field, the t2g orbitals occur at lower energy than the eg orbitals. Only important for odd number occupancy of the eg level Î eg1 or eg3. The effect of Jahn-Teller distortions is best documented for Cu2+ complexes (with 3 electrons in the eg level) where the result is that most complexes are found to have elongation along the z-axis. Tetragonally distorted complexes: the Jahn-Teller Effect • Consider the octahedral Cu2+ ion Î d9 Î t2g6 eg3 • The eg levels are degenerate Î 3 electrons Î dx2-y2 1 dz2 2 or dx2-y2 2 dz2 1 • Hence the degeneracy of the eg levels is lifted Î ligands along the axes experience different shielding effects Î 2 e-s vs. 1 e-. • Resulting in z -axis contraction (dx2-y2 2 dz2 1) or elongation (dx2-y2 1 dz2 2). As the z axis elongation increases Î the energy of the dz2 orbital drops lower than the dxy orbital which along with dx2-y2 orbital rises in energy. X' X X Cu X 2+ X X' X = Cl X = Br X=F Cu---X 2.30 2.40 1.93 Cu---X΄ 2.95 3.18 2.27 Tetragonally distorted complexes: the Jahn-Teller Effect •(dx2-y21 dz2 2) due to the 2 electrons on the z axis the dz2 orbital is more shielded from the Cu2+ centre Î elongation along this axes Î also called tetragonal distortion Î lower in energy. Also arises due to ligand dissimilarity, e.g. in MA4B2 Δ Very rare Tetragonally distorted complexes: the Jahn-Teller Effect • The distortion has resulted in lower energy for the system Î stabilisation. • Therefore many Cu2+ complexes are tetragonal even with six identical ligands, e.g. [Cu(H2O)6]2+. • The main reason for tetragonal distortion is to remove the instability brought about by the non-linearity and achieve stability in TM complexes. • No distortion for t2g3, t2g3 eg2 , t2g6, t2g6 eg2 , t2g6 eg4 . •Important cases of distortion are: t2g3 eg1 (high spin Cr2+ and Mn3+) and t2g6 eg1 (low spin Co2+ and Mn3+) Tetragonally distorted complexes: the Jahn-Teller Effect dx2-y2 dx2-y 2 dx2-y 2 eg dz 2 dz 2 dxy dz 2 dxy t2g dxz dyz dxy dxz dyz dxz dyz (a) Octahedral field (b) Small tetragonal distortion (c) Large tetragonal distortion Square planar complexes Recall Jahn-Teller tetragonal distortion discussed above. Mostly formed by metals with d8 ions with strong field ligands, e.g. [NiII(CN)4]2-. Note that [NiIIX4]2- forms tetrahedral complexes, why? All complexes of Pt(II) and Au(III) are square-planar, including those with weak field ligands such as [PtCl4]2-. Also true for most complexes of Rh(I), Ir(I) and Pd(II) Î 4d and 5d complexes. z y dx2-y2 dxy dz2 } dxz; dyz x A square planar complex is obtained when it is energetically favourable to have the configuration dyz2dxz2dxy2dx2-y22 for 4d8 and 5d8 ions. Full dz2 and empty dx2-y2 Î Molecular Orbital Theory (MOT) and Ligand–Field Theory (LFT) • The purely e-static approach of the CFT makes it unsuitable to adequately explain metal-ligand bonding. TM compounds such as Ni0(CO)4 with Ni in zero oxidation state must be purely covalent Î no M-L e-static attraction. • We have already seen that on purely e-static grounds, the order of the spectrochemical series is unviable, e.g. we expect CO < H2O < C2O42- < EDTA4- Î this is however not the case. • Evidence from NMR & ESR Î unpaired e-n density on the ligands Î sharing of e-ns Î strong evidence for M-L covalency. •LFT is the theory that combines CFT with covalency (MOT) • Essentially LFT is able to give an understanding of the true origins of Δo and the spectrochemical series by taking into account the roles of σ- and π- bonding in TM chemistry. Oh σ bonding z z y x + y + + + + + x + s z z y + y + x x - - Pz z z y y - - x + + x Px z z y y + + x x - Py z - - x - + dz2 x + - y y - - y + y + z z + z + dx2-y2 x + - + x t2g: Non bonding Orbitals Remain localised on metal atom Oh σ bonding In LFT the building up principle is used in conjunction with a molecular energy level diagram constructed from metal orbitals and symmetry adapted linear combination of atomic orbitals (SALC). For an octahedral geometry: Six SALC with σ symmetry interact with the metal orbitals The orbitals of the central metal atom divide by symmetry into 4 sets, viz: Singly degenerate s Triply degenerate p Doubly degenerate d Triply degenerate d a1g t1u eg t2g Typical molecular energy levels diagram of an octahedral complex showing the frontier orbitals in the tinted box Oh σ bonding • The ligand-field model for octahedral TM complexes such as Co(NH3)63+ and CoF63- assumes that the 3d, 4s, and 4p orbitals on the metal overlap with one orbital on each of the six ligands to form a total of 15 molecular orbitals. • Six of these orbitals are bonding molecular orbitals, whose energies are much lower than those of the original atomic orbitals. • Another six are antibonding molecular orbitals, whose energies are higher than those of the original atomic orbitals. • Three are best described as nonbonding molecular orbitals, because they have essentially the same energy as the 3d atomic orbitals on the metal. • Note: The d-orbitals of LFT are the molecular orbitals and not the atomic orbitals of the CFT • Summary: Oh σ bonding • The allocation of electrons is similar to CFT •12 ligand electrons are accommodated in the bonding orbitals. • metal electrons are accommodated in the NB (t2g) and AB (eg*) orbitals. • The separation between the frontier orbitals (Δo) is similar to CFT. • The type of complex that is obtained will also depend on Δo and P. • The origin of Δo and the spectrochemical series are clearer: • good σ-donor ligands (e.g. CH3- and H-) result in strong metalligand overlap Î larger value of Δo due to a more strongly AB eg*. •Except for ligands in which there are no orbitals of π symmetry available for bonding Î σ-bonding only ligands, such as CH3- and Hπ-bonding is the main determinant of Δo and the spectrochemical series for TM complexes. Oh σ bonding Q. Construct molecular orbital energy level diagrams for CoIII ion in the following octahedral crystal fields: 1. Low spin 2. High spin Clearly show: • the relative positions of the metal, ligand and complex orbitals • 10Dq • placement of electrons in each case Answer: Oh σ bonding Steps for determining d-orbital energy level diagrams for TM complexes: • Determine the oxidation state of the metal • Determine the number of d electrons • Determine ligand field Îstrong or weak Î strong field = low spin Î weak field = high spin • Draw the energy level diagram Oh σ bonding Antibonding MOs 4p Δο 4s Six donor orbitals 6NH3 each donating 2 e-s 3d 2 x2-y2 z xy xz yz NB MOs Bonding MOs Atomic orbitals in metal ion Molecular Orbital diagram for [CoIII(NH3)6 ]3+ Molecular orbitals Atomic orbitals in ligand ion Oh σ bonding Antibonding MOs 4p Δο 4s Six donor orbitals 6 F- each donating 2 e-s 3d 2 x2 -y2 z xy xz yz NB MOs Clearly good σ donor ligand Result in good M-L overlap Î Strongly antibonding eg set Atomic orbitals in metal ion Molecular Orbital diagram for CoF63- Bonding MOs Molecular orbitals Atomic orbitals in ligand ion Ligand–Field Theory: π bonding π bonding is important for ligands with orbitals of local π symmetry with respect to the M-L axis Î e.g. metal dxy and ligand py orbitals. These interaction with the previously NB metal t2g orbitals Î influences Δo. π-donor ligand Î has filled orbitals of π symmetry Î These are π-bases, e.g. Cl-, Br-, OH- and H2O. These have L→M e-n donation. π-acceptor ligand Î has empty π orbitals that are available for occupation Î These are π acids, e.g. CO and PR3 ligands Î capable of M→L back donation to form π- bonds. Summary of influence of π-bonding on Δo: π-donor ligands decrease Δo. π-acceptor ligands increase Δo. π-donor ligands decrease Δo. π-acceptor ligands increase Δo. Ligand–Field Theory: π bonding Summary: strong σ- or π-donor Î weak field ligands. π acceptor ligands higher in E than t2g. π donor ligands lower in E than t2g. π-acceptors Î strong field ligands. Increasing Δo → π donor < weak π donor < no π effects < π acceptor e.g. I-, Br- Cl- FH2O NH3 PR3, CO